Tumor-agnostic treatment, or the drug-based treatment of tumors, regardless of anatomic origin, based on the agent’s molecular alteration target, has been a widely studied potential treatment paradigm for patients with limited disease-directed treatment options. Pembrolizumab was the first of 5 currently US Food and Drug Administration (FDA)-approved tumor-agnostic therapies (ie, pembrolizumab, larotrectinib, entrectinib, selpercatinib, and dostarlimab) and 1 tumor-agnostic combination therapy (ie, dabrafenib plus trametinib) for any solid tumor that has progressed after previous treatment.1-6 These tumor-agnostic therapies have shown sustained objective response rates in a variety of tumors,1-6 supporting this approach as a future direction for cancer treatment.
Based on these promising data, this study investigated the tumor-agnostic potential of poly (ADP-ribose) polymerase (PARP) inhibitors. The DNA replication process is unstable and is susceptible to alterations.7 These alterations are a result of exposure to environmental agents, byproducts of normal cellular metabolism, and spontaneous disruption of chemical bonds in the DNA. These alterations may cause transcription of nonfunctional proteins that limit the survivability of the cell. Cells repair these alterations by several methods, including base excision repair, nucleotide excision repair, mismatch repair, homologous recombination repair (HRR), and nonhomologous end joining.7
PARP is an enzyme involved in base excision repair and consists of 2 subdomains: helical domain and adenosine diphosphate ribosyltransferase (ART) catalytic domain.7 When DNA is altered, PARP binds to single-stranded breaks, which nullifies the helical domain’s autoinhibitory function and activates the catalytic function of ART. This catalytic activity leads to the generation of poly (ADP-ribose) chains on target proteins, which promotes the recruitment DNA repair effectors.7
The oncogenic loss of DNA repair effectors (eg, breast cancer susceptibility proteins, BRCA1, and BRCA2) causes cancer cells to become overly dependent on the PARP-related single-strand break repair pathway instead of the reliable double-strand repair mechanism via HRR involving functional BRCA1 or BRCA2.7
By inhibiting PARP, a bimodal lethal effect occurs for tumor cells with a BRCA1 or a BRCA2 mutation.7 PARP inhibition forces the cancer cell to use nonhomologous end joining to repair DNA damage, which is a highly error-prone pathway that produces nonfunctional proteins and a nonviable cancer cell.7,8
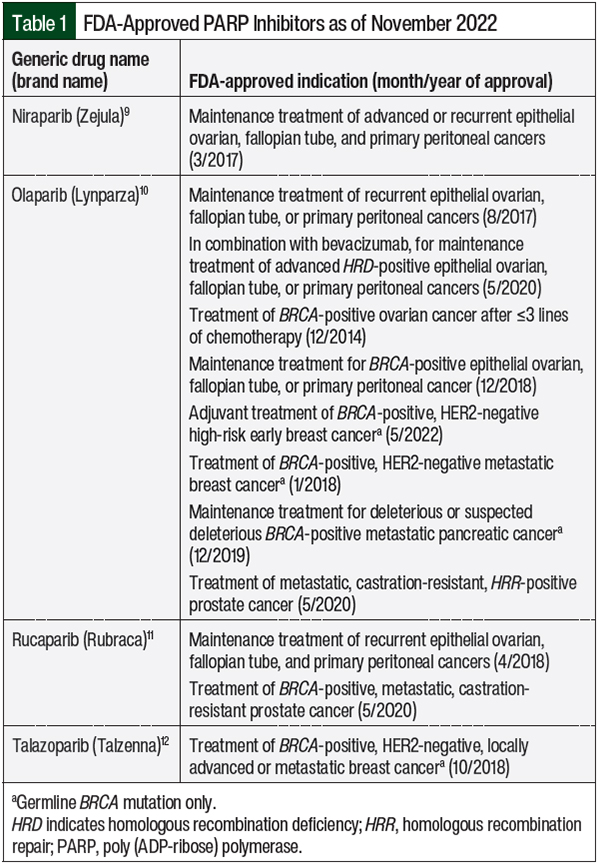
PARP inhibitors suppress the catalytic activity of ART and are FDA-approved for the treatment of various types of cancer associated with BRCA1 or BRCA2 mutations (Table 1).9-12
In vitro and limited in vivo clinical trials have shown that PARP inhibitors may have a lethal effect on cancer cells via the inhibition of HRR caused by mutations besides BRCA1 and BRCA2.13,14
Olaparib and niraparib have proved effective in the treatment of epithelial ovarian, fallopian tube, and primary peritoneal cancers and vague homologous recombination deficiency (HRD) mutations, for which they are FDA approved.9,10 However, the anticancer effect of the majority of PARP inhibitors in vague HRD and HRR mutations has only been noted in limited cases of small-cell lung cancer, pancreatic cancer, and sarcomas.15
In vitro studies have since sought to identify predictive genetic biomarkers for response to PARP inhibitors. The deficiency of RAD51, RAD54, DSS1, RPA1, NBS1, ATR, ATM, CHK1, CHK2, FANCD2, FANCA, and FANCC are a few of many DNA damage-signaling proteins known to induce sensitivity to PARP inhibition in vitro.14 Thus, it is prudent to gather data on any and all relevant mutations that result in DNA repair enzyme deficiencies exhibited by tumors to assess fully the possible tumor-agnostic treatment with PARP inhibitors.
The purpose of this study was to analyze the use of PARP inhibitors for off-label conditions and to evaluate the patient outcomes. Pertinent tumor mutations were also gathered in an effort to try and correlate the response to treatment with PARP inhibitor use in patients with certain tumor mutations and to identify the tumor-agnostic effectiveness of the PARP inhibitors.
Methods
This retrospective chart review was conducted at Huntsman Cancer Institute at the University of Utah Health in Salt Lake City, UT. The study included patients aged ≥18 years who received at least 1 dose of a PARP inhibitor for an off-label use between January 2015 and September 2020. Patients who were pregnant, incarcerated, participating in a clinical trial, or were receiving olaparib for the treatment of metastatic pancreatic cancer with BRCA mutation after December 2019 or for HRR-positive, metastatic, castration-resistant prostate cancer after May 2020 were excluded. Electronic health records (EHRs) and insurance denial records were used to identify the patients for study enrollment.
This study was deemed exempt by the local Institutional Review Board.
The primary outcome was PARP inhibitor–related overall survival, defined as the time from the PARP inhibitor prescription start date, corroborated with the provider-documented start date (whichever was later), until the patient’s death or last known follow-up. The secondary outcomes were PARP inhibitor–related progression-free survival, defined as the time from the initiation of a PARP inhibitor to the discontinuation of the PARP inhibitor, as determined by provider documentation, or until the next line of treatment started that did not include a PARP inhibitor; patients’ adverse events compared with the FDA-indicated adverse events; and the impact of non-BRCA mutations on the patients’ outcomes.
The data collection via manual chart review of the EHR included the patients’ baseline characteristics, malignancy, tumor tissue mutations (excluding variants of unknown significance), line(s) of treatment, and concomitant treatment (if applicable); the providers’ documentation of response; the Common Terminology Criteria for Adverse Events16 grade of adverse events; and if PARP inhibitor–related supportive care was used. The providers’ documentation of response was used instead of the RECIST (Response Evaluation Criteria in Solid Tumors) criteria, because of insufficient retrospective data necessary to determine whether the response criteria were met. Descriptive statistics, such as percentages, mean, median, and range, were used in this study because of the small sample size.
Results
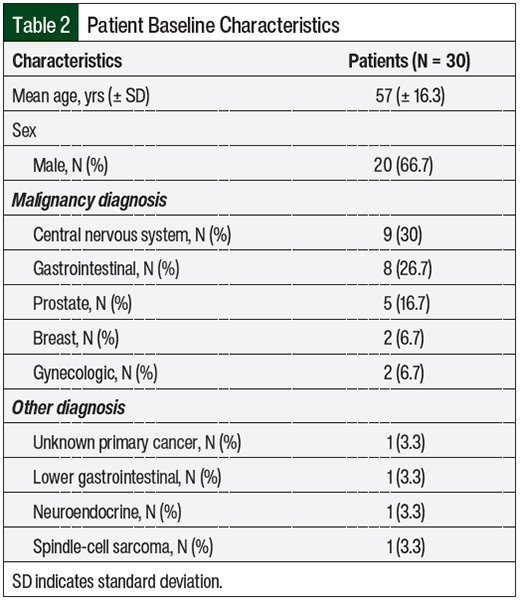
A total of 86 patients were originally identified, and 30 of them met the study’s inclusion criteria. The mean patient age was 57 years, and approximately 67% of the patients were men (Table 2).
PARP inhibitors were the first, second, or third line of treatment for 40% of the patients; fourth, fifth, or sixth line for 53.3% of the patients; and seventh line or greater for 6.7% of the patients. A majority of the patients received olaparib (76.6%), with the remaining patients receiving talazoparib (16.7%), niraparib (6.7%), or rucaparib (3.3%). One patient received 2 sequential PARP inhibitors (olaparib followed by talazoparib). In all, 20% of the patients received treatment in combination with either bevacizumab or carboplatin.
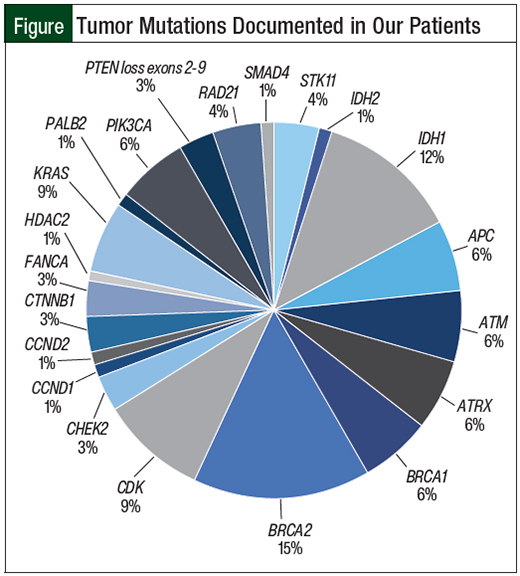
There was an average of 4 tumor mutations per patient (range, 1-8); all documented mutations are listed in the Figure.
In the olaparib cohort, 4 patients had anaplastic astrocytoma, 4 had anaplastic oligodendroglioma, 1 had glioblastoma, 1 had cervical carcinoma, 3 had colon cancer, 1 had pancreatic cancer, 1 had ampullary carcinoma, 1 had unknown primary cancer, 1 had spindle-cell sarcoma, 5 had prostate cancer, and 1 had breast cancer.
In the niraparib cohort, 1 patient had endometrial cancer and 1 had breast cancer. The 1 patient in the rucaparib cohort had pancreatic cancer.
Finally, in the talazoparib cohort, the diagnoses were extrahepatic cholangiocarcinoma, metastatic squamous-cell carcinoma of the esophagus, unknown primary cancer (the same patient who previously received olaparib in this study), and neuroendocrine tumor.
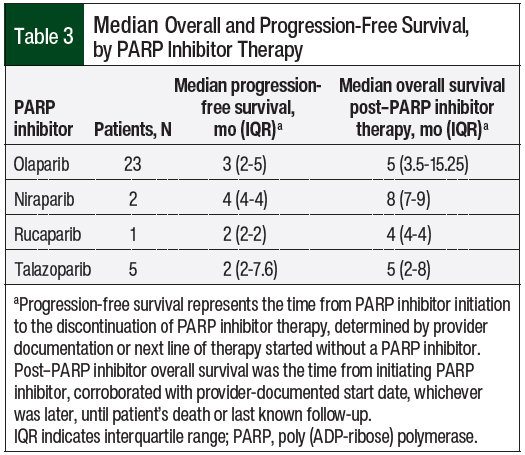
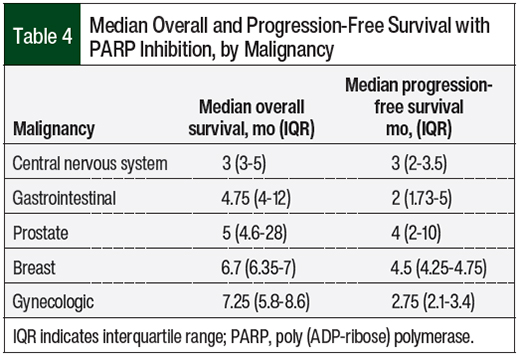
The median combined PARP inhibitor–related overall survival and progression-free survival were 5 months (interquartile range [IQR], 4-11) and 3 months (IQR, 2-4.5), respectively. The overall survival and progression-free survival data by PARP inhibitor and disease state are shown in Table 3 and Table 4, respectively.
A total of 20 patients had adverse events after receiving PARP inhibitor treatment. The adverse events were patient-reported or were determined by objectively measured values. The most common adverse events were fatigue (29.6%), nausea (18.5%), thrombocytopenia (14.8%), anemia (14.8%), abdominal pain (7.4%), diarrhea (3.7%), dizziness (3.7%), hypertension (3.7%), and neutropenia (3.7%). In all, 7 patients had grade 3 or 4 adverse events, including thrombocytopenia (N = 2), anemia (N = 2), dizziness (N = 1), neutropenia (N = 1), and hypertension (N = 1).
As a result of these adverse events, there were 9 (30%) dose reductions, which were caused by thrombocytopenia, anemia, and neutropenia; 6 dose adjustments were associated with olaparib therapy, 2 were associated with talazoparib, and 1 with niraparib.
Discussion
Tumor-agnostic therapy is expanding with the use of programmed cell death 1 inhibitors and neurotrophic tyrosine receptor kinase inhibitors. Because of limited in vitro and pending clinical trials exploring the tumor-agnostic potential of PARP inhibitors, our goal was to analyze the outcomes, safety, and tumor mutations of patients receiving PARP inhibitors for the treatment of various solid tumors at our institution.
The study patients had generally poor prognosis and limited therapeutic options, and PARP inhibitor therapy was among the last lines of treatment for them. Therefore, the goal of treatment was symptom improvement, reduction of disease progression, and prolongation of life.
The median overall survival and progression-free survival were similar among the patients with different malignancies. The adverse-event profile of PARP inhibitors in this heterogeneous population aligned with the adverse events reported in the published literature at relatively similar rates—approximately 36% for olaparib, 65% for talazoparib, and 72% for niraparib.9,10,12
There were variances in overall survival and progression-free survival with combinations of tumor mutations, but no correlations were identified between certain mutation combinations and improved overall survival or progression-free survival.
Cancer is caused by multiple genetic mutations, but it is often difficult to determine if a combination of mutations, and which specific combination, is responsible for a patient’s tumor and its proliferation.17 If a particular patient’s cancer has multiple driver mutations that are susceptible to targeted therapies, it may be reasonable to consider innovative combinations of targeted therapies.
In an attempt to further elucidate the complexity of tumor mutations, we collected molecular-sequencing information about the study population. These data, however, did not reveal any certain combination of tumor mutations that rendered a relatively improved response to PARP inhibitors.
Within the patients’ mutations, 6 (RAD51, FANCA, ATM, ATRX, PALB2, and CHEK2) are associated with the notoriously complex BRCA repair pathway,18 which poses the possibility of expanding potential drug targets.
When assessing the combination of mutations, it was difficult to identify which ones were exquisitely sensitive to PARP inhibitors. For example, the patient with the longest progression-free survival (48 months) and overall survival (48 months) received off-label treatment with olaparib for spindle-cell sarcoma. That patient’s tumor mutations were TAF1 truncation, TP53, BRCA1, amplification of KIT, PGDFRA, RICTOR, and FGF10.
The patient with the second longest progression-free survival (24 months) and overall survival (24 months) received off-label treatment with talazoparib for extrahepatic cholangiocarcinoma. This patient’s tumor mutations were BRCA1, STK11 loss exon 1, CDK6 amplification, MYC amplification, ACVR1B, MAP2K4, RAD21 amplification, and TP53.
Both of these patients exhibited the BRCA1 mutation, which may have contributed to their prolonged progression-free survival and overall survival response.
Limitations
Our study has several limitations, including its retrospective design, its adverse-effect data based on provider documentation, and the inability to determine patient adherence. The retrospective design of the study meant that the data available to collect were limited to what was documented, which might have varied by patient, tumor type, or provider style. Therefore, data collected such as adverse effects may be incomplete.
In addition, patient adherence to treatment may affect the outcome data collected in this study, which may limit our results.
Having a single-institution data set, a small sample size, and the lack of a comparator cohort are additional limitations. A larger sample size from multiple diverse institutions and including a comparator cohort would increase the likelihood of attaining statistical significance for the off-label use of PARP inhibitors.
Conclusion
This study did not include statistical analysis, because of its small sample size; therefore, we cannot conclude that PARP inhibitor therapy significantly improves overall survival and progression-free survival in a tumor-agnostic treatment approach. However, the patients in this study received PARP inhibitors after multiple previous therapies and gained a median of 3 months of overall survival, which is clinically significant for such patients with limited treatment options.
This study shows that the tumor-agnostic use of PARP inhibitors for the treatment of solid tumors with relevant mutations is promising and may prolong the overall survival of this patient population with limited therapy options.
Author Disclosure Statement
Dr Wasef, Dr Fritz, and Dr Cavalieri have no conflicts of interest to report.
References
- US Food and Drug Administration. FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication. May 23, 2017. www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication. Accessed November 11, 2022.
- US Food and Drug Administration. FDA approves larotrectinib for solid tumors with NTRK gene fusions. December 14, 2018. www.fda.gov/drugs/fda-approves-larotrectinib-solid-tumors-ntrk-gene-fusions. Accessed November 11, 2022.
- US Food and Drug Administration. FDA approves entrectinib for NTRK solid tumors and ROS-1 NSCLC. August 16, 2019. www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc. Accessed November 11, 2022.
- US Food and Drug Administration. FDA grants accelerated approval to dostarlimab-gxly for dMMR advanced solid tumors. February 1, 2022. www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors. Accessed November 11, 2022.
- US Food and Drug Administration. FDA grants accelerated approval to dabrafenib in combination with trametinib for unresectable or metastatic solid tumors with BRAF V600E mutation. June 23, 2022. www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid. Accessed November 11, 2022.
- US Food and Drug Administration. FDA approves selpercatinib for locally advanced or metastatic RET fusion-positive non-small cell lung cancer. September 21, 2022. www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-non-small-cell-lung. Accessed November 11, 2022.
- Yi M, Dong B, Qin S, et al. Advances and perspectives of PARP inhibitors. Exp Hematol Oncol. 2019;8:29. doi: 10.1186/s40164-019-0154-9.
- Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol. 2011;22:268-279.
- Zejula (niraparib) capsules, for oral use [prescribing information]. GlaxoSmithKline; September 2022. https://gskpro.com/content/dam/global/hcpportal/en_US/Prescribing_Information/Zejula_Capsules/pdf/ZEJULA-CAPSULES-PI-PIL.PDF. Accessed November 11, 2022.
- Lynparza (olaparib) tablets, for oral use [prescribing information]. AstraZeneca Pharmaceuticals; October 2022. https://den8dhaj6zs0e.cloudfront.net/50fd68b9-106b-4550-b5d0-12b045f8b184/00997c3f-5912-486f-a7db-930b4639cd51/00997c3f-5912-486f-a7db-930b4639cd51_viewable_rendition__v.pdf. Accessed November 11, 2022.
- Rubraca (rucaparib) tablets, for oral use [prescribing information]. Clovis Oncology; June 2022. https://clovisoncology.com/pdfs/RubracaUSPI.pdf. Accessed November 11, 2022.
- Talzenna (talazoparib) capsules, for oral use [prescribing information]. Pfizer; September 2021. https://labeling.pfizer.com/ShowLabeling.aspx?id=11046. Accessed November 11, 2022.
- Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917-921.
- McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109-8115.
- Pilié PG, Gay CM, Byers LA, et al. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25:3759-3771.
- US Department of Health & Human Services. Common Terminology Criteria for Adverse Events (CTCAE). Version 5.0. November 27, 2017. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf. Accessed July 8, 2022.
- Al Hajri Q, Dash S, Feng WC, et al. Identifying multi-hit carcinogenic gene combinations: scaling up a weighted set cover algorithm using compressed binary matrix representation on a GPU. Sci Rep. 2020;10:2022. doi: 10.1038/s41598-020-58785-y.
- Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12:68-78.