Thrombotic microangiopathy (TMA) refers to changes in the walls of arterioles and capillaries, which predisposes them to microvascular thrombosis.1 Hemolytic uremic syndrome (HUS) is a subset of TMA that is characterized by nonimmune microangiopathic hemolytic anemia, thrombocytopenia, and organ damage, especially to the kidneys in the form of acute kidney injury (AKI).2 HUS is most often preceded by diarrhea (often bloody) that results from enteric infections by Shiga toxin–producing bacteria (Shigella dysenteriae and some strains of Escherichia coli, such as serotypes O157:H7 and O104:H4) and is labeled as ST-HUS or typical HUS.2 Recent diarrheal illness with bloody stools can imply ST-HUS, but it can also be a manifestation of bowel ischemia secondary to TMA of any other cause.3 The key distinguishing factor is the timing of AKI in relation to the occurrence of diarrhea.4 In ST-HUS, the diarrheal illness typically precedes AKI by several days.4 The term atypical HUS (aHUS) was used for non–Shiga-toxin–mediated HUS, which is caused by the inappropriate activation of the alternative complement pathway resulting from inherited mutations in complement regulatory genes or by autoantibodies directed against complement regulatory proteins (complement factor H [CFH], complement factor I [CFI], membrane cofactor protein [MCP; CD46]).5 Hence, aHUS can also be referred to as complement-mediated TMA (CM-TMA).
aHUS is a rare disorder with an annual incidence of 2 cases per million population in the United States.6 Approximately 50% to 60% of complement-mediated HUS results from gene mutations with reported relative frequencies of 20% to 30% from CFH or CFH-related (CFHR) proteins, 5% to 15% from CD46/MCP, 4% to 10% from CFI, 2% to 10% from C3, 1% to 4% from complement factor B, and 3% to 5% from thrombomodulin genes.7-9 More than 100 mutations of CFH have been reported, most of which are missense mutations that do not affect the levels of CFH and C3.10 The results of a 2007 study that assessed outcomes based on the genotypes of 46 children showed that patients with CFH mutations have poor prognosis, with most patients progressing to end-stage renal disease (ESRD) or death within 1 year of presentation, whereas mutations in CD46/MCP do not result in disease progression to ESRD, although relapse is common.11 The results of a study in 2013 of 214 French patients showed that clinical outcomes, such as the incidence of ESRD and mortality, were not different from disease that resulted from different genotypes in adults versus in children, although CFH mutations were associated with a significantly poorer prognosis in children versus in adults.12
Although many genetic forms of aHUS pathology are known, only some of the carriers precipitate the disease. The reason is unclear, but it is postulated that there is an antecedent trigger event that is thought to play a role in complement activation, such as immunologic disorders, systemic diseases, drug administration, or infections such as influenza A and B, especially H1N1 strain; cytomegalovirus; Bordetella pertussis; and COVID-19.13,14
Before the era of eculizumab and ravulizumab, plasma exchange was the mainstay of management for aHUS. In an Italian study, 48% of children and 67% of adults with aHUS had ESRD or died during the first episode of aHUS or within 3 years of its onset.7 In the previously mentioned French study, 29% of children and 56% of adults with aHUS required renal replacement therapy or died within 1 year of follow-up.12 Eculizumab is effective in resolving and preventing TMA, improving renal function and hematologic outcomes, and reducing morbidity and mortality.15-17 Ravulizumab is engineered from eculizumab resulting in molecules that target the same complement protein with a longer half-life.18,19
Ravulizumab and eculizumab are available only through a restricted program under a Risk Evaluation and Mitigation Strategy in the United States.17,19 Although the treatment of aHUS with eculizumab is started with a loading dose of 900 mg weekly for 4 weeks followed by a maintenance dose of 1200 mg at week 5 and every 2 weeks thereafter,17 treatment with ravulizumab requires only 1 loading dose of 900 to 3000 mg and a maintenance dose of 2100 to 3600 mg every 8 weeks.19 Along with a reduced dosing frequency, ravulizumab introduces a cost-savings benefit as a result of its longer half-life versus eculizumab.17-19 Most previous studies of aHUS and the restrictive use of eculizumab have been described in European patients.12,20-22 There is a paucity of data in North American patients.
Methods
This institutional review board–approved, retrospective, observational cohort study was conducted at the University of Arkansas for Medical Sciences, Little Rock, AR, between January 1, 2013, and January 31, 2021. The data were collected by an extensive review of the electronic medical records of patients who were diagnosed with aHUS and received eculizumab in the Department of Hematology and Oncology. We were able to identify 21 patients who met the study inclusion criteria, including 4 patients who were lost to follow-up, for a total of 17 patients included in the study. The inclusion criteria included the presence of microangiopathic hemolytic anemia with thrombocytopenia, age ≥18 years, and ADAMTS13 (disintegrin and metalloprotease with thrombospondin-1–like motif, member 13) activity ≥10%. The study exclusion criteria were TMA associated with HUS, scleroderma renal crises, or antiphospholipid syndrome, and being lost to follow-up (ie, not being seen in the clinic until January 31, 2021). The pertinent patient demographics, such as age, sex, ethnicity, and clinical information, including presenting features, examination findings, laboratory results, complications, and treatment instituted, were collected. We also studied the outcomes for mortality and laboratory or clinical evidence of relapse in the patients who stopped treatment with eculizumab or who switched from treatment with eculizumab to ravulizumab.
In this study, AKI was defined as an increase in serum creatinine ≥0.3 mg/dL in the past 48 hours or ≥1.5 times baseline, which is known or presumed to have occurred in the past 7 days, or urine volume <0.5 mL/kg per hour for 6 hours based on the Kidney Disease: Improving Global Outcomes guidelines. Chronic kidney disease (CKD) was defined as the presence of kidney damage (typically urinary albumin excretion of ≥30 mg/day or equivalent) or decreased kidney function (estimated glomerular filtration rate [eGFR] <60 mL/min/1.73 m2) for 3 or more months. ESRD is defined as eGFR <15 mL/min/1.73 m2 with the signs and symptoms of uremia, such as nausea, vomiting, and pericarditis, with a requirement of dialysis.
Results
The mean age at diagnosis was 47.4 ± 18 years (range, 23-82 years). The female to male ratio was 4.6:1 (Table 1).
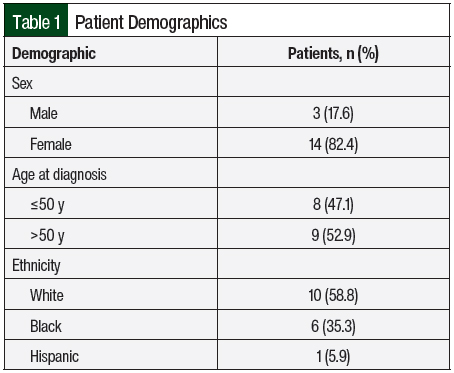
The most prevalent extrarenal presentation was elevated blood pressure (>140/90 mm Hg) followed by central nervous system symptoms (altered mental status and seizures) and gastrointestinal symptoms (nausea/vomiting and diarrhea; Figure 1). In the 2 patients who had diarrhea, it was nonbloody and did not precede, but was concurrent with, the illness. In our study, 2 (11.8%) patients had previous TMA and 3 (17.6%) patients had a previous kidney transplant. Of the 3 patients with kidney transplant, 2 were receiving immunosuppression with mycophenolate mofetil and tacrolimus. A total of 2 (11.8%) patients had a history of cancer, one of whom had multiple myeloma and received chemotherapy with bortezomib, dexamethasone, cisplatin, doxorubicin, cyclophosphamide, and etoposide (VD-PACE) plus melphalan and was undergoing autologous stem cell transplant, and the other patient had ampullary cancer and was receiving gemcitabine.
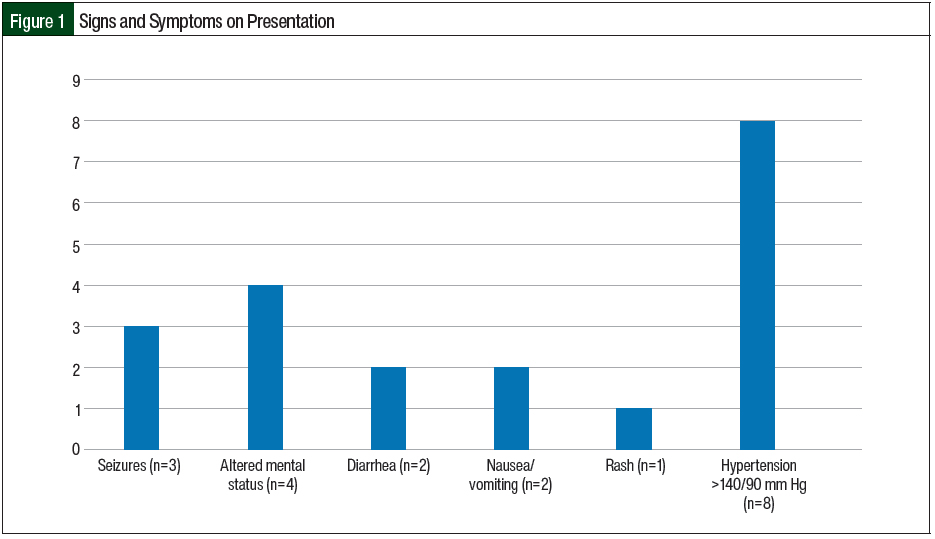
Infection and pregnancy (n=4; 23.5% each) were the most frequent complement-amplifying conditions in the study (Figure 2). For 2 patients, it was their first pregnancy and for the other 2 patients, it was their second and third pregnancies. The patients presented at the second-, sixth-, and sixteenth-week postpartum visits, respectively.
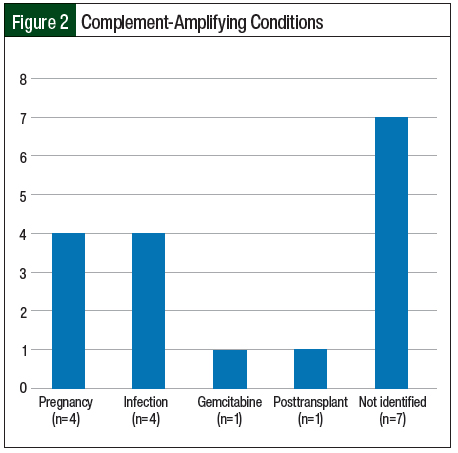
All of the study patients except 1 had AKI and thrombocytopenia at presentation (n=16; 94.1% each; Table 2). In all, 8 (47.1%) patients were asymptomatic and had abnormal laboratory findings that led to a diagnosis of aHUS. Complement genetic testing was done in 100% of the study population. In all, 8 (47.1%) patients in our study had an identifiable gene mutation (Table 3). The most observed pathogenic mutations in the study patients were CFH and CFHR1/3. One of the patients with a positive mutation panel—heterozygous mutation in CFHR3, variant of unknown significance in CFH, and CD46 mutations—had 2 sisters with aHUS.
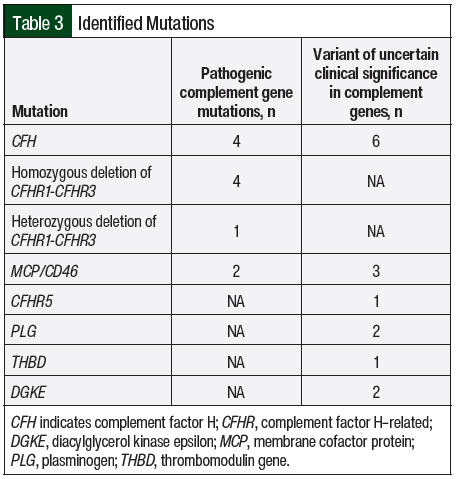
A total of 14 (82.2%) patients received therapeutic plasma exchange, 2 (11.8%) patients received intravenous (IV) immunoglobulin, and 4 (23.5%) patients received IV methylprednisolone 1 mg/kg before receiving C5 inhibitors (Table 4). A total of 17 patients received eculizumab per the standard dosing. In all, 5 (29.4%) patients opted to completely stop drug therapy, and 4 (23.5%) patients chose to switch from eculizumab to ravulizumab because of the ease of treatment duration (every 8 weeks with ravulizumab vs every 2 weeks for eculizumab). All of these 9 patients had stable renal and hematologic parameters with no signs of disease relapse (Table 5).
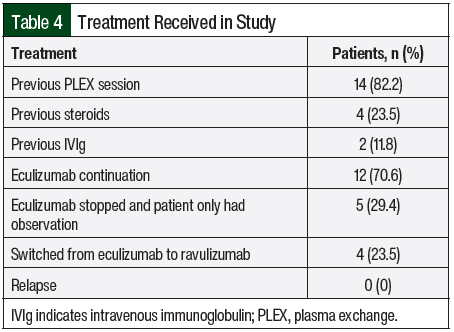
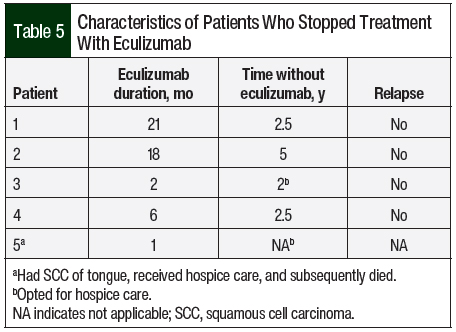
All of the study patients were vaccinated against meningococcus. The patients whose vaccination status could not be ascertained or who were unvaccinated on the initiation of C5 inhibitors were vaccinated as soon as possible and received antimicrobial prophylaxis for up to 2 weeks after vaccination. A total of 10 (58.8%) patients received antimicrobial prophylaxis with penicillin, and 1 patient had an allergy to penicillin and hence received prophylaxis with levofloxacin. One patient who received penicillin prophylaxis had disseminated cryptococcal infection.
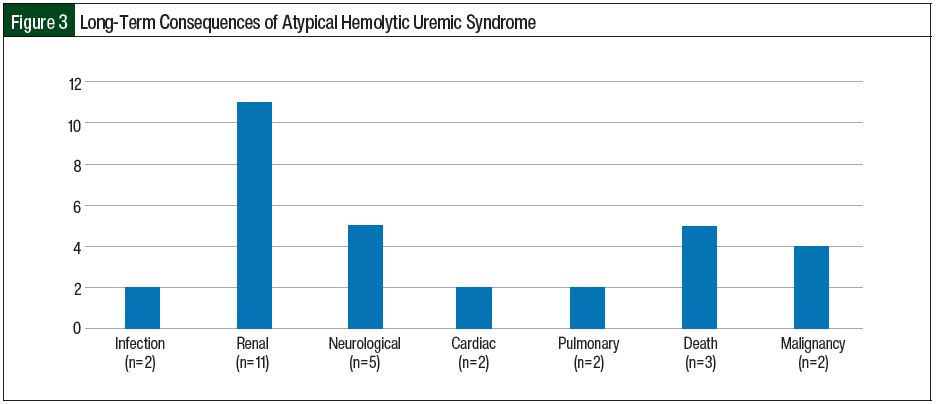
All of the postpartum women in our study were able to breastfeed while receiving eculizumab with no long-term complications in the neonates. One patient had 2 subsequent deliveries without ante-, intra-, or postpartum consequences or repeat triggering of aHUS. In all, 3 patients died in our case series, including 1 patient of E coli gram-negative sepsis with acute respiratory distress syndrome at diagnosis, 1 patient of complications related to multiple myeloma 5 years after being diagnosed with CM-TMA, and 1 patient who had squamous cell carcinoma of the tongue with subsequent transition of care to hospice 2 years after being diagnosed with aHUS. A total of 11 (64.7%) patients had CKD, with 5 (29.4%) patients requiring temporary dialysis and 6 (35.3%) requiring permanent dialysis after disease progression to ESRD. In all, 3 (17.6%) patients had continuing seizures and had to receive antiepileptic agents, 2 (11.8%) pregnant patients had cardiomyopathy, and 2 (11.8%) patients had pulmonary complications (pneumonia and pulmonary hypertension; Figure 3). Two (11.8%) patients had cancer (colonic adenocarcinoma and squamous cell carcinoma of the tongue) 5 years and 2 years, respectively, after being diagnosed with aHUS.
Discussion
All of our study patients except for 1 had AKI as the initial presentation. The other extrarenal manifestations in our study included hypertension (n=8), altered mental status (n=4), seizures (n=3), diarrhea (n=2), nausea/vomiting (n=2), and rash (n=1). The median age of presentation in our study was 47 years and most patients were women, which correlates with a 2015 study by Sperati and Moliterno, in which the median age of presentation in adults with TMA and AKI was 46 years and most patients were women.23 The penetrance of this disease is low, but the disease risk is higher in carriers of causative variants and is not equally distributed in generations, with siblings and offspring of the patients having a much higher disease risk than parents.24 As reported in 2 studies, family history of HUS is obtained in approximately 20% to 30% of patients.2,12 Only 1 of our patients with a positive mutation panel had a family history of aHUS, having 2 sisters with aHUS. In our study, 4 (23.5%) patients had pregnancy as a trigger for aHUS. Other triggers included infection (n=4), autologous stem cell transplant for multiple myeloma after receiving VD-PACE plus melphalan (n=1), and receiving gemcitabine (n=1) for the treatment of ampullary carcinoma. We were not able to identify any triggers in 7 (41%) of our patients.
Similar to a study of 308 patients with aHUS in the United Kingdom,25 genetic mutations were detected in 8 (47%) of our study patients with the most often identified mutation being homozygous or heterozygous deletion in CFHR1/3 (n=5), followed by splice-site, nonsense, missense mutations in different exons of CFH (n=4). Although several studies have reported that aHUS associated with CFH mutations presents during infancy or early childhood,11,25-29 the previously mentioned French study showed that 25 of 59 (42%) patients with CFH mutations initially presented between the ages of 20 years and 40 years, which was similar to our study.12 As in other studies,9,30-32 a significant number of patients with CM-TMA in our study had have mutations in more than 1 complement protein.
Diagnosing aHUS is challenging, especially when differentiating aHUS from other forms of TMAs. In the majority of patients, thrombotic thrombocytopenic purpura will be a consideration. Two of our patients had thrombotic thrombocytopenic purpura and aHUS with 10% ADAMTS13 activity and a positive mutation panel (CFHR1/R3), but the patients’ disease did not improve after plasma exchange or treatment with steroids. Six (35%) patients had a mild-to-moderate reduction in ADAMTS13 activity (>10%-70%). This reduction is in concordance with a study by Feng and colleagues who concluded that partial ADAMTS13 deficiency may occur in patients with complement-mediated HUS and should always be evaluated in patients with complement-mediated aHUS.33
Treatment with C5 inhibitors should be started as soon as possible, ideally within the first 48 hours of admission, based on the data from the studies showing tremendous improvement in outcomes including ESRD and death.15,21,34-36 Serious infections with Neisseria species (other than Neisseria meningitidis) and other encapsulated bacteria have been reported. All patients should receive N meningitidis, Streptococcus pneumoniae, and Haemophilus influenza type b vaccination. In a report from the Centers for Disease Control and Prevention, 16 cases of meningococcal disease were identified in eculizumab recipients in the United States between 2008 and 2016.37 One patient in our study had disseminated cryptococcal infection. Other major adverse events related to treatment with eculizumab include hypertension, influenza, peritonitis, bacteriuria, and venous sclerosis at infusion sites.17,38
The optimum duration of eculizumab treatment in patients with aHUS has not been determined. Given the high cost of eculizumab (average sale price of 10 mg is $225.79 as per medicare part B),39 the possibility of having disseminated meningococcal infections or immune-mediated reactions, and the unknown effects of long-term therapy, lifelong therapy with eculizumab is an ongoing debate among international hematologists. Discontinuing eculizumab therapy has been described in extremely limited experiences in some observation studies.20,40-42 In our study, 5 (29.4%) patients who opted to completely stop their drug therapy had been in remission based on hematologic and renal parameters. Case series by Ardissino and colleagues (N=16), Wijnsma and colleagues (N=20), and Chaturvedi and colleagues (N=31) support the discontinuation of eculizumab therapy, with 11, 13, and 17 patients, respectively, being in remission.20,42,43 In a prospective, observational study of 93 patients by Menne and colleagues, there was a potential risk for additional TMA events and renal function decline after the discontinuation of treatment with eculizumab, especially in the patients with genetic or autoimmune complement abnormalities.22 A review by Macia and colleagues on the discontinuation of treatment with eculizumab by 52 patients in published case reports, 61 of 130 patients in clinical trials, and 76 of 296 patients in the aHUS Registry, of whom 16, 12, and 12 patients, respectively, had TMA events, concluded that the risk for TMA after the discontinuation of eculizumab treatment is unpredictable in severity and timing.44 In addition, some studies have reported that risk of relapse after eculizumab is genetically geared with higher relapse rates in CFH, MCP, CFI, and CD46 but this genetic correlation with relapse was not observed in our study.40,41
According to a study by Rondeau and colleagues, ravulizumab is noninferior to eculizumab across all efficacy end points in patients with previously treated or C5 inhibitor–naïve aHUS.18 In a study by Wang and colleagues, ravulizumab provided lifetime per-patient cost reductions (discounted) versus eculizumab of 32.4% and 35.5% in adults and pediatric base cases, respectively.45 The total costs of ravulizumab versus eculizumab were $12,148,748 and $17,979,007, respectively, for adults and $11,587,832 and $17,959,814, respectively, for children.45 In our study, 4 (23.5%) patients chose to switch from treatment with eculizumab to ravulizumab because of the ease of therapy duration (every 8 weeks with ravulizumab rather than every 2 weeks with eculizumab). This reduction in the frequency of treatments can ensure better compliance and quality of life for patients and can prevent complications related to recurrent IV access. More studies are needed on the assessment of the restricted use of eculizumab and the clinical use, benefits, and safety profile of ravulizumab.
A total of 11 (64.7%) patients in our study had CKD, with 6 (35.3%) patients requiring permanent dialysis after progression to ESRD. One of the pregnant patients had 2 subsequent pregnancies without recurrence. Three patients died in our study. Hence, there was no significant correlation between the patients’ genotype and phenotype.
Limitation
The small sample size of the study is a major limitation.
Conclusion
The clinical diagnosis of aHUS can be challenging, especially with extrarenal manifestations. A female preponderance for aHUS was seen, and the majority of patients had complement-gene mutations. No ante-, intra-, or postpartum complications occurred in current or subsequent pregnancies among patients with aHUS triggered by pregnancy who received C5 inhibitors. No correlation was found between renal outcomes and genotypes.
The decision to discontinue or to switch treatment from eculizumab to ravulizumab will likely decrease healthcare costs and improve patient compliance, safety, and relapse rates. Larger multicenter studies or trials are needed to further validate this study’s findings.
Author Disclosure Statement
Dr Sasapu is on the Speaker’s Bureau of BMS, ADC Therapeutics, Alnylam Pharmaceuticals, and Gilead, and owns stocks in Crisper Therapeutics, Alnylam Pharmaceuticals, Apple, Google, Legend Biotech, Gracell Biotech, and Gilead; Dr Amisha, Dr Konda, Dr Malik, Mr Fugere, and Dr Roy have no conflicts of interest to report.
References
- George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med. 2014;371(7):654-666.
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035-1050.
- George JN. Clinical practice. Thrombotic thrombocytopenic purpura. N Engl J Med. 2006;354(18):1927-1935.
- Tarr PI, Gordon CA, Chandler WL. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365(9464):1073-1086.
- Leon J, LeStang MB, Sberro-Soussan R, et al. Complement-driven hemolytic uremic syndrome. Am J Hematol. 2023;98(suppl 4):S44-S56.
- Constantinescu AR, Bitzan M, Weiss LS, et al. Non-enteropathic hemolytic uremic syndrome: causes and short-term course. Am J Kidney Dis. 2004;43:976-982.
- Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859.
- Bu F, Zhang Y, Wang K, et al. Genetic analysis of 400 patients refines understanding and implicates a new gene in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018;29(12):2809-2819.
- Geerdink LM, Westra D, van Wijk JA, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol. 2012;27(8):1283-1291.
- Martín Merinero H, Zhang Y, Arjona E, et al. Functional characterization of 105 factor H variants associated with aHUS: lessons for variant classification. Blood. 2021;138(22):2185-2201.
- Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al; for the French Society of Pediatric Nephrology. Differential impact of complement mutations on clinical characterstics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18:2392-2400.
- Fremeaux-Bacchi V, Fakhouri F, Garnier A, et al. Genetics and outcomes of atypical hemolytic uremic syndrome: a nationwide French series comparing children and adults. Clin J Am Soc Nephrol. 2013;8:554-562.
- Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676-1687.
- Rodríguez de Córdoba S. aHUS: a disorder with many risk factors. Blood. 2010;115(2):158-160.
- Legendre CM, Licht C, Muus P, et al. Terminal complement inhibitor eculizumab in atypical hemolytic–uremic syndrome. N Engl J Med. 2013;368:2169-2181.
- Rondeau E, Cataland SR, Al-Dakkak I, et al. Eculizumab safety: five-year experience from the global Atypical Hemolytic Uremic Syndrome Registry. Kidney Int Rep. 2019;4:1568-1576.
- Soliris (eculizumab) injection, for intravenous use [prescribing information]. Alexion Pharmaceuticals; November 2020. https://alexion.com/Documents/Soliris_USPI.pdf. Accessed August 17, 2023.
- Rondeau E, Scully M, Ariceta G, et al; for the 311 Study Group. The long-acting C5 inhibitor, ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020;97:1287-1296. Errata in: Kidney Int. 2020;98:1621; Kidney Int. 2021;99:1244.
- Ultomiris (ravulizumab-cwvz) injection, for intravenous or subcutaneous use [prescribing information]. Alexion Pharmaceuticals; July 2022. https://alexion.com/Documents/Ultomiris_USPI.pdf. Accessed August 17, 2023.
- Wijnsma KL, Duineveld C, Volokhina EB, et al. Safety and effectiveness of restrictive eculizumab treatment in atypical haemolytic uremic syndrome. Nephrol Dial Transplant. 2018;33:635-645.
- Ardissino G, Salardi S, Colombo E, et al. Epidemiology of haemolytic uremic syndrome in children. Data from the North Italian HUS network. Eur J Pediatr. 2016;175:465-473.
- Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20:125.
- Sperati CJ, Moliterno AR. Thrombotic microangiopathy: focus on atypical hemolytic uremic syndrome. Hematol Oncol Clin North Am. 2015;29:541-559.
- Ardissino G, Longhi S, Porcaro L, et al. Risk of atypical HUS among family members of patients carrying complement regulatory gene abnormality. Kidney Int Rep. 2021;6(6):1614-1621.
- Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115:379-387.
- Caprioli J, Bettinaglio P, Zipfel PF, et al; for the Italian Registry of Familial and Recurrent HUS/TTP. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297-307.
- Neumann HP, Salzmann M, Bohnert-Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. 2003;40(9):676-681.
- Landau D, Shalev H, Levy-Finer G, et al. Familial hemolytic uremic syndrome associated with complement factor H deficiency. J Pediatr. 2001;138(3):412-417.
- Warwicker P, Donne RL, Goodship JA, et al. Familial relapsing haemolytic uraemic syndrome and complement factor H deficiency. Nephrol Dial Transplant. 1999;14(5):1229-1233.
- Goodship TH, Cook HT, Fakhouri F, et al; Conference Participants. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91(3):539-551.
- Esparza-Gordillo J, Jorge EG, Garrido CA, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. 2006;43(11):1769-1775.
- Kavanagh D, Richards A, Fremeaux-Bacchi V, et al. Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2007;2(3):591-596.
- Feng S, Eyler SJ, Zhang Y, et al. Partial ADAMTS13 deficiency in atypical hemolytic uremic syndrome. Blood. 2013;122:1487-1493.
- Christmann M, Hansen M, Bergmann C, et al. Eculizumab as first-line therapy for atypical hemolytic uremic syndrome. Pediatrics. 2014;133(6):e1759-1763.
- Greenbaum LA, Fila M, Ardissino G, et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016;89(3):701-711.
- Muff-Luett M, Sanderson KR, Engen RM, et al. Eculizumab exposure in children and young adults: indications, practice patterns, and outcomes-a Pediatric Nephrology Research Consortium study. Pediatr Nephrol. 2021;36(8):2349-2360.
- McNamara LA, Topaz N, Wang X, et al. High risk for invasive meningococcal disease among patients receiving eculizumab (Soliris) despite receipt of meningococcal vaccine. MMWR Morb Mortal Wkly Rep. 2017;66:734-737.
- Licht C, Greenbaum LA, Muus P, et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015;87:1061-1073.
- Centers for Medicare and Medicaid Services (CMS). ASP Pricing Files. www.cms.gov/medicare/payment/fee-for-service-providers/part-b-drugs/average-drug-sales-price/asp-pricing-files. Accessed September 9, 2023.
- Fakhouri F, Fila M, Provôt F, et al. Pathogenic variants in complement genes and risk of atypical hemolytic uremic syndrome relapse after eculizumab discontinuation. Clin J Am Soc Nephrol. 2017;12(1):50-59.
- Fakhouri F, Fila M, Hummel A, et al. Eculizumab discontinuation in children and adults with atypical hemolytic-uremic syndrome: a prospective multicenter study. Blood. 2021;137(18):2438-2449.
- Ardissino G, Testa S, Possenti I, et al. Discontinuation of eculizumab maintenance treatment for atypical hemolytic uremic syndrome: a report of 10 cases. Am J Kidney Dis. 2014;64(4):633-637.
- Chaturvedi S, Dhaliwal N, Hussain S, et al. Outcomes of a clinician-directed protocol for discontinuation of complement inhibition therapy in atypical hemolytic uremic syndrome. Blood Adv. 2021;5:1504-1512.
- Macia M, de Alvaro Moreno F, Dutt T, et al. Current evidence on the discontinuation of eculizumab in patients with atypical haemolytic uraemic syndrome. Clin Kidney J. 2017;10:310-319.
- Wang Y, Johnston K, Popoff E, et al. A US cost-minimization model comparing ravulizumab versus eculizumab for the treatment of atypical hemolytic uremic syndrome. J Med Econ. 2020;23:1503-1515.