Background: Glioblastoma multiforme (GBM) is a highly malignant glial tumor characterized by rapid growth and angiogenesis. Current frontline therapy consists of surgical resection, radiation, and chemotherapy; however, all patients will progress. Over the past decade, there have been increases in the quality and quantity of clinical data regarding the treatment of patients with GBM.
Objective: The purpose of this review is to describe the pathways of tumorigenesis, review relevant data in both the frontline and recurrent disease settings, and discuss the place in therapy of novel treatment options for GBM.
Methods: Stupp and colleagues revolutionized the management of patients with GBM with their 2005 landmark study that demonstrated the benefits of surgery and radiotherapy plus concomitant and adjuvant temozolomide. Other studies have shown that after disease progression, the administration of bevacizumab alone and in combination with cytotoxic chemotherapy resulted in increased progression-free survival. Data also support the use of daily temozolomide in recurrent disease, leading to similar results, although there are no comparative studies with bevacizumab and temozolomide.
Conclusion: Much progress has been made in the treatment of GBM. Despite these advances, nearly all patients progress after frontline therapy and options remain limited.
Glioblastoma multiforme (GBM) is classified as a grade 4 central nervous system (CNS) tumor by the World Health Organization and is the most malignant of the glial tumors. GBM is characterized by rapid mitotic activity, infiltrative growth, and necrosis. Microvascular proliferation is often present in the tumor and suggestive of aggressive angiogenesis. Patients may present with a variety of neurologic symptoms, including headaches, seizures, confusion, memory loss, personality changes, and focal neurologic deficits. Magnetic resonance imaging is usually confirmatory, showing an enhancing mass, peritumoral edema, and central areas of necrosis. Multi modality treatment of GBM typically includes surgery, radiation, and chemotherapy. The natural history of the disease is progression, and prognosis remains poor, with a survival rate of <15 months for a majority of patients.1
Epidemiology According to the Central Brain Tumor Registry of the United States, in 2010 there were 22,020 new diagnoses and 13,140 deaths attributed to primary malignant brain and CNS tumors.2 GBM comprises 60% to 70% of all newly diagnosed glioma, occurs at a median age of 64 years, and is more common in men than in women.1 GBM has been associated with rare familial syndromes that introduce genomic instability; however, the only proved environmental risk factor associated with the development of GBM is exposure to high-dose radiation, although a history of chemotherapy has also been associated.3,4
The most common chemotherapies associated with the development of secondary GBM have been antimetabolite therapies (methotrexate and 6-mercaptopurine) for the treatment of acute lymphoblastic leukemia.4 Genetic polymorphisms affecting detoxification, DNA repair, and cell cycle regulation have also been implicated in tumorigenesis.5 The incidence of GBM has been increasing over the past 2 decades, largely because of improvements in imaging, availability of medical care, and treatment options for elderly patients and reclassification of brain tumors.5
Molecular Pathogenesis
GBM is described clinically based on tumorigenesis. GBM is classified into 2 main subtypes based on biologic and genetic differences, with malignant transformation ultimately resulting from genetic abnormalities and dysregulation of cell-signaling pathways. Primary (de novo) GBM is now recognized to be a molecular phenotype separate from the slower-growing secondary GBM that evolves from lower-grade gliomas.
Primary GBM is common in patients aged >50 years and is characterized by epidermal growth factor receptor (EGFR) overexpression and mutation, including variant EGFR amplification, loss of heterozygosity of chromosome 10q leading to the deletion of phosphatase and tensin homolog (PTEN), and p16 deletion.5,6
Secondary GBM is less common and arises from tumors with p53 mutations. It is characterized by overexpression of the platelet-derived growth factor receptor (PDGFR), aberrations in p16 and retinoblastoma pathways, and loss of heterozygosity of chromosome 10q.5,6
Despite these differences, primary and secondary tumors are morphologically identical and are treated with similar regimens, although there may be differences in response in patients receiving targeted therapies. Growth factor receptor signaling involving EGFR and PDGFR result in the activation of pathways, such as the Ras-MAP kinase pathway, involved in cell cycle progression and cell proliferation, and the PI3K/Akt pathway, which results in inhibition of apoptosis and cellular proliferation.5,6
PTEN, which is located on chromosome 10 and is a regulator of the PI3K/Akt pathway, is inactivated in approximately 50% of patients with GBM. Vascular endothelial growth factor (VEGF) and activation of the VEGF receptor (VEGFR) are also upregulated, leading to angiogenesis and tumor survival. Finally, there is some evidence that neural stem cells can give rise to gliomas, thereby secreting VEGF and promoting angiogenesis within the tumor microenvironment.6,7
View larger version
First-Line Treatment Options
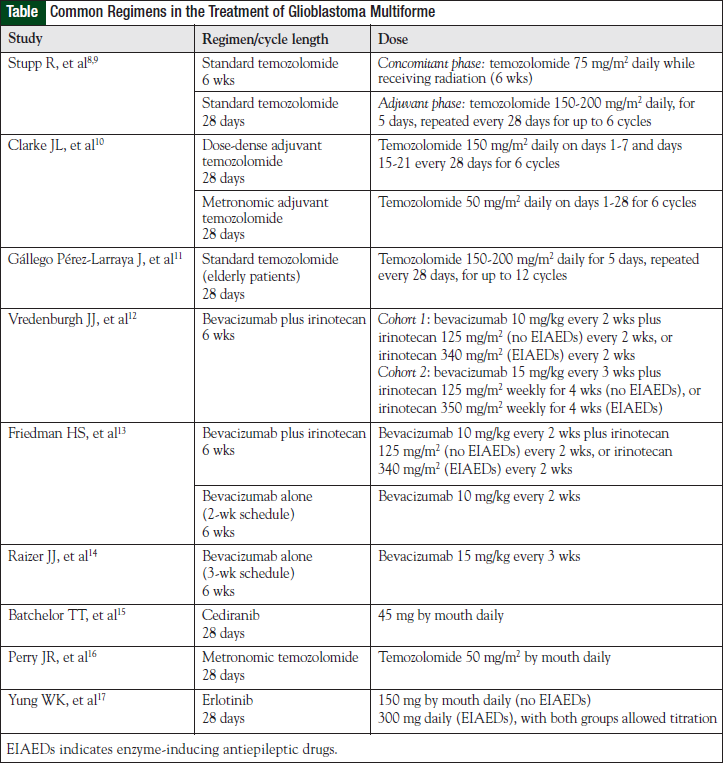
The most effective first-line treatment of GBM to date remains optimal surgical resection, followed by the combination of concomitant daily temozolomide and postoperative radiation, followed by 6 cycles of adjuvant temozolomide (see Table for selected chemotherapy regimens and dosing information).8-17 When compared with postoperative radiation alone, temozolomide-based therapy resulted in a survival increase of 2.5 months (12.1 vs 14.6 months, respectively) or a 37% relative reduction in the risk of death, with a median follow-up of 28 months.8 Although this represents a significant improvement, temozolomide-based therapy had a 2-year survival rate of only 26.5%.7 With 5 years of follow-up, this statistic is even more dismal at only 9.8%.9
Significant room for improvement remains for first-line treatment options. It is also important to consider using prophylaxis for Pneumocystis carinii pneumonia when temozolomide is given in the concurrent phase, because of the increased risk for opportunistic infections secondary to potential severe lymphocytopenia.
An important marker identified in the study by Hegi and colleagues was MGMT (O6-methylguanine-DNA methyltransferase).18 The MGMT gene encodes for a DNA repair protein, which removes alkyl groups from the O6 position of guanine; this is an important site for DNA alkylation. In tumor cells with high levels of MGMT, resistance can develop through blunting the effect of alkylating agents, such as temozolomide and dacarbazine. MGMT silencing via promoter methylation results in a loss of MGMT function, leaving cells more susceptible to alkylating drugs. In fact, in this temozolomide trial, MGMT promoter methylation was an independent favorable prognostic marker. In patients whose tumors had MGMT promoter methylation and received temozolomide- based therapy, the median overall survival (OS) was 21.7 months and the 2-year OS rate was 46%.18
Although MGMT is clearly predictive of outcome in patients with GBM who receive temozolomide-based therapy, the clinical benefit of this marker remains to be determined.19 This is mainly a result of the lack of effective therapeutic alternatives in patients whose MGMT promoter methylation status does not suggest a significant benefit from alkylating drug–based therapy. Several investigators are searching for possible dosing alterations that may overcome this resistance mechanism.
Clarke and colleagues evaluated the safety and efficacy of 2 alternative adjuvant temozolomide dosing schedules in the frontline management of 85 patients with GBM.10 The first strategy was metronomic or continuous daily dosing of temozolomide. This method of delivery was postulated to provide combined antitumor, as well as antiangiogenic, effects via damage to endothelial cells in tumor vasculature. In addition, metronomic delivery of continuous low-dose temozolomide results in inhibition of MGMT that could possibly overcome this major resistance pathway. The second delivery schedule was dose-dense temozolomide, which is based on the Norton-Simon model of cell proliferation. This method also has the potential to inhibit MGMT. Both methods were well tolerated, with no unexpected toxicities or rates of toxicities observed. Lymphopenia was common but did not result in any cases of P carinii pneumonia. Two-year survival in the dose-dense and metronomic arms were 34.8% and 28%, respectively.10 Although this trial was limited by the small sample size and lack of direct comparison to standard-dose temozolomide, the tolerable side-effect profile and impressive efficacy of dose-dense and metronomic temozolomide warrant further investigation in randomized controlled trials.10
Another strategy to overcome chemotherapy-resistant mechanisms is the use of multidrug chemotherapy. The drug that has generated recent optimism for the treatment of GBM is bevacizumab, an anti-VEGF monoclonal antibody. This optimism has mostly been generated from data with bevacizumab in the treatment of GBM refractory to first-line temozolomide-based therapy. With its unique mechanism of action, as well as a manageable toxicity profile, bevacizumab is ideal to investigate with other antineoplastic drugs. In fact, a large international, phase 3, multicenter, randomized trial is currently under way to evaluate standard temozolomidebased therapy, with or without the addition of bevacizumab. 20 The Radiation Therapy Oncology Group has initiated a multicenter, phase 3 trial investigating the use of bevacizumab as first-line therapy.21
A preliminary safety report from a separate pilot phase 2 trial included bevacizumab, which was added to standard, temozolomide-based therapy.22 Although relatively high rates of fatigue, myelosuppression, wound breakdown, and thrombosis were observed, the rates of these toxicities were deemed acceptable, and enrollment in the trial continues. The use of the combination of temozolomide and bevacizumab should be limited to clinical trials until further results confirming the safety and efficacy of this combination are available.22
With so much focus on systemic therapy for GBM, it is easy to forget the benefits demonstrated with local therapy with carmustine wafers. Placebo-controlled trials of carmustine wafers implanted at the time of initial surgery demonstrated a significant survival benefit in favor of carmustine wafers. Patients were randomized to receive either carmustine wafers plus radiotherapy or identical-appearing placebo wafers plus radiotherapy.23 A total of up to 8 wafers were implanted in each patient.
Systemic therapy was prohibited for patients with GBM unless recurrence was documented. Of the 240 patients enrolled in the trial, 207 had GBM. The median survival was 13.5 months in the carmustine group and 11.4 months in the placebo group, with 1-year survival rates of 59.2% and 49.6%, respectively.23 This resulted in a 31% (95% confidence interval [CI], 3%-51%) risk reduction of death in the carmustine-treated group compared with the placebo-treated group, which was significant in the GBM subgroup (P = .04). The time to Karnofsky performance status (KPS) and neuroperformance deterioration also favored the carmustine group. The adverse event profiles were similar for carmustine and placebo.23
Paucity of Data for the Elderly
A GBM population group not addressed in many clinical trials is the elderly. For example, the landmark temozolomide trial excluded patients aged >70 years.8 The paucity of literature for elderly patients with GBM has resulted in a lack of clear guidance on the best treatment modalities for this population.
Radiation therapy offers benefit over supportive care alone in elderly patients with a good performance status. In a randomized trial conducted by the Association of French-Speaking Neuro-Oncologists, patients with GBM aged >70 years with a KPS >70 were randomized to supportive care only or supportive care and radiotherapy (focal radiation in daily fractions of 1.8 Gy given 5 days weekly, for a total dose of 50 Gy).24 A total of 85 patients were enrolled at 10 institutions. The trial was discontinued after the first interim analysis as a result of superior survival rate in the radiotherapy arm. At a median follow-up of 21 weeks, the hazard ratio for death in the radiotherapy arm was 0.47 (95% CI, 0.29-0.76; P = .002), which yielded a median survival benefit of 12.2 weeks. The median survival for patients receiving radiotherapy plus supportive care was 29.1 (95% CI, 25.4- 34.9) weeks, and for those receiving supportive care only, 16.9 (95% CI, 13.4-21.4) weeks. The KPS and cognition declined over time; however, there was no difference between the 2 groups. No serious adverse events were reported with radiotherapy.24
Although elderly patients were not included in the trial by Stupp and colleagues,8,9 temozolomide has been studied in a phase 2 trial of patients with GBM aged ≥70 years. In this nonrandomized trial, temozolomide 150 to 200 mg/m2 daily for 5 days every 4 weeks was evaluated.11 Radiotherapy was not administered in this trial. Of note, only patients with a poor performance status (KPS <70) were included in this trial.11
A total of 70 patients from 8 institutions were enrolled in the trial. Median treatment duration with temozolomide was 2 cycles per patient (range, 0-13 cycles). Dose delays and dose reductions occurred in 20% and 24% of patients, respectively. The median OS was 25 weeks (95% CI, 19-28 weeks). The 6-month and 12-month OS rates were 44.3% (95% CI, 32.7%-55.9%) and 11.4% (95% CI, 3.9%-18.8%), respectively. Temozolomide was generally well tolerated. Grade 3 to 4 neutropenia and thrombocytopenia occurred in 13% and 14% of patients, respectively.11
Recurrent Disease Treatment
The majority of patients with disease recurrence are not eligible for further irradiation or surgical intervention. Median survival is 25 weeks, and survivorship at 1 year is estimated to be approximately 25%.25-27 Progression-free survival (PFS) is correlated with OS in this patient population, and the PFS at 6 months is between 9% and 15%.25-27 GBM is one of the most vascularized of all tumors, and angiogenesis is critical to disease progression. Proangiogenic factors, such as VEGF, regulate vascularity, and VEGF is overexpressed in areas of necrosis, hypoxia, and endothelial proliferation. VEGF expression is correlated with tumor activity and aggressiveness, with GBM having the highest levels of all gliomas.28,29
High expression of VEGF is associated with microvascular proliferation, accelerated tumor expansion, and poor outcomes, but these tumors are also the most likely to respond to antiangiogenic therapy. It is hypothesized that a higher VEGF ligand-to-receptor ratio may indicate more persistent VEGFR activation, and this ratio may be increased with age.28,29
Bevacizumab is a humanized immunoglobulin G1 monoclonal antibody that binds to and neutralizes circulating VEGF. Bevacizumab has been evaluated as a single drug and in combination with cytotoxic chemotherapy in patients with recurrent GBM.
Stark-Vance was the first to publish promising results of bevacizumab in combination with irinotecan.30 Previously, irinotecan had been used as single-drug therapy in patients with recurrent GBM because of good CNS penetration; however, response rates were less than 20%, with many patients not responding at all.31-33 The topoisomerase 1 inhibitor irinotecan works via a different mechanism of action than alkylation and is not affected by MGMT, is not highly protein bound, and readily crosses the blood–brain barrier, making it a logical choice for combination with bevacizumab. Twentyone patients with high-grade gliomas (53% GBM) were treated with bevacizumab and irinotecan.30 In the earlier study, 43% of patients had an objective response, and among those not meeting criteria for response, most had radiographic improvement consisting of reductions in peritumoral edema and contrast enhancement.30 These early results indicated that bevacizumab may have a role in the management of high-grade gliomas.30
Vrendenburgh and colleagues verified the beneficial results of bevacizumab in combination with irinotecan in 35 patients with histologically proven GBM that progressed after external-beam radiation therapy and concurrent temozolomide.12 The trial included 2 cohorts of patients receiving differing doses and schedules of bevacizumab and irinotecan, with the irinotecan dose dependent on the presence of enzyme-inducing antiepileptic drugs.12
Response to therapy was determined by magnetic resonance imaging (MacDonald Criteria) and clinical examination. Six-month PFS was 46% and median OS was 42 weeks after 68 weeks of follow-up. A total of 57% of patients had at least a partial response to therapy, and 7 patients completed 1 year of therapy. Thirteen patients stopped therapy as a result of disease progression, and 11 patients stopped therapy because of toxicity. Other reasons for discontinuation included venous thromboembolism (VTE), proteinuria, and CNS hemorrhage.12
Friedman and colleagues further established the role of bevacizumab in a multicenter, phase 2, noncomparative trial of bevacizumab alone and in combination with irinotecan.13 In the group that received bevacizumab alone versus the group that received irinotecan plus bevacizumab, objective responses were 28.2% and 37.8%, PFS rates at 6 months were 42.6% and 50.3%, and median OS rates were 9.2 months and 8.7 months, respectively.13 The study did not achieve power to detect significant differences between these groups. Patients receiving bevacizumab alone achieved overall response rates and PFS rates at 6 months that are greater than historical rates achieved (which have been <20%) with cytotoxic chemotherapy alone. This demonstrates the value of the addition of antiangiogenic therapy in recurrent GBM.
This begs the question whether the additional benefits of adding cytotoxic chemotherapy to bevacizumab are worth the risks when the response rates are similar between the 2 groups. Comparative studies of bevacizumab alone and bevacizumab in combination with cytotoxic chemotherapy are needed to confirm the synergy between the drugs in the recurrent setting. Toxicities common to bevacizumab (ie, hypertension, proteinuria, delayed wound healing, and hemorrhage) were similar between the groups. There were increased incidences of VTE, diarrhea, and hematologic toxicity in the group receiving irinotecan.13
Currently, bevacizumab is recommended to be dosed on a schedule of every 2 weeks in patients with GBM. A phase 2 evaluation of bevacizumab dosing every 3 weeks produced 6-month PFS results that were more favorable than historical controls14; however, no studies comparing dosing strategies of bevacizumab have been performed to make formal recommendations on dose density.
Cediranib is an oral pan-VEGFR tyrosine kinase inhibitor (TKI) that also has activity against PDGF-β and c-Kit. Based on its mechanism of action and potential molecular targets, cediranib should have activity in primary and in secondary GBM. Preliminary studies showed that edema decreased and tumor vasculature was normalized after treatment with cediranib.15 Cediranib was further evaluated in 31 patients with recurrent GBM in a single-center phase 2 study with a primary end point of PFS at 6 months. All patients had undergone surgery and radiation and most (29 of 31) had initial treatment with temozolomide. The 6-month PFS rate was 25.8%, median PFS was 117 days, and OS was 227 days. The use of cediranib also allowed decreased dexamethasone doses. The most common toxicities were diarrhea, fatigue, and hypertension. There were no reports of intracranial hemorrhage.15
Temozolomide has been the backbone of initial treatment of GBM. However, patients relapse and, for patients receiving chemotherapy, a resting period is required for nontumor cell recovery to occur. It is hypothesized that during this time DNA repair may take place, allowing for tumor regrowth. When compared with procarbazine, temozolomide in patients at first relapse resulted in an improved 6-month PFS rate (21% vs 8%; P = .008), and this freedom from disease progression also was associated with maintained health-related quality of life.34 Continuous low-dose or metronomic scheduling of temozolomide has been attempted to suppress MGMT activity, increase dose intensity, and increase the antiangiogenic effects of other chemotherapies.16
The RESCUE study tested this hypothesis and examined the effects of daily temozolomide when given to patients with high-grade glioma or GBM at first progression after exposure to conventional dosing.16 Patients with GBM were stratified into 3 groups according to their previous duration of treatment with temozolomide and time of progression. All patients received temozolomide 50 mg/m2 daily continuously for up to 12 months or until disease progression. Overall 6-month PFS rate for patients with GBM was 23.9%, with 7.4% progressing while receiving extended adjuvant temozolomide beyond 6 cycles but before completion of adjuvant treatment having the shortest time to progression (1.8 months; P = .027) and 14.8% having the shortest survival at 1 year. Best responses were in the early- or late-progression groups. Lymphopenia was the most common toxicity, and there was no incidence of P carinii pneumonia. The 6-month PFS rate and time to progression were similar between patients with methylated and unmethylated MGMT promoter status.16
Other Therapeutic Options
EGFR has emerged as a potential target in patients with malignant glioma, and overexpression of EGFR is associated with a poor prognosis in GBM. Response rates to small-molecule TKIs remain low and results are not uniform, although there is a subset of patients with EGFRvIII and PTEN expression that appear to benefit from TKI therapy.35
Erlotinib has been evaluated in multiple phase 2 studies for high-grade glioma in patients receiving and not receiving enzyme-inducing antiepileptic drugs. Best responses obtained to date have been 6-month PFS rates of 20%, which are still higher than historical rates of 9% to 15%; however, none of the studies are controlled studies. Because stable disease is the most common response in patients receiving small-molecule therapy, it appears that erlotinib is cytostatic in GBM.17 Erlotinib has also been evaluated in combination with bevacizumab in a phase 2 study of patients with recurrent malignant glioma; however, the 6-month PFS rate was lower (29.2%) among patients with GBM compared with other bevacizumab salvage regimens.36 The most common side effects in studies with erlotinib were diarrhea and rash, which both correlate with increased PFS in GBM.17,36 Molecular stratification of patients with GBM may identify those likely to respond to therapy.35
Patients who do not have an intact PTEN may have increased response to mTOR inhibitor and EGFR TKI combinations. Some evidence of clinical response has been seen in phase 1 investigations involving peptide vaccines37 and single drugs such as bortezomib38; however, additional studies need to be performed to determine the benefit of these drugs.
Conclusion
GBM is the most aggressive of the glial tumors. Much progress has been made over the past decade in GBM research, and a new standard of care has emerged, with nearly all patients receiving radiotherapy plus concomitant and adjuvant temozolomide for newly diagnosed GBM. Despite these advancements, all patients will progress eventually, and most patients are not eligible for additional radiation or for surgical resection of tumor on disease progression. Chemotherapy and biotherapy remain the standard of care for these patients.
Because of the angiogenic potential of GBM, bevacizumab has emerged as a promising therapy for use in the recurrent disease setting. Six-month PFS, which tracks with OS, is a useful end point in this patient population, and treatment with bevacizumab alone and in combination with cytotoxic chemotherapy have improved 6- month PFS over single-drug alkylator therapy.
It is still unknown if the benefits of bevacizumab in combination with cytotoxic chemotherapy outweigh the risks, and further comparative trials are needed to assess the value of this combination. Combinations of bevacizumab and chemotherapy may result in improved PFS, but these regimens are associated with increased toxicities, including myelosuppression and thromboembolic events, which may limit their use in some patient populations.
Data have emerged that temozolomide therapy can be repeated for patients with relapsed disease, and that continuous metronomic dosing may be beneficial, by extending PFS without further decreasing quality of life with increased toxicity.
Questions still remain about the optimal timing for bevacizumab therapy, and the best schedule for it. Based on the best data available, bevacizumab should still be recommended to be given every 2 weeks to patients who have recurrent disease. This decade has brought much improvement and insight to the management of GBM; however, there is still a lot to learn and much progress to be made.
Author Disclosure Statement
Dr Buie and Dr Valgus have reported no conflicts of interest.
References
- Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492- 507.
- Central Brain Tumor Registry of the United States. Fact sheet. www.cbtrus.org/factsheet/factsheet.html. Accessed February 1, 2012.
- Edick MJ, Cheng C, Yang W, et al. Lymphoid gene expression as a predictor of risk of secondary brain tumors. Genes Chromosomes Cancer. 2005;42:107-116.
- Relling MV, Rubnitz JE, Rivera GK, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. Lancet. 1999;354:34-39.
- Schwartzbaum JA, Fisher JL, Aldape KD, Wrensch M. Epidemiology and molecular pathology of glioma. Nat Clin Pract Neurol. 2006;2:494-503.
- Nicholas MK, Lukas RV, Chmura S, et al. Molecular heterogeneity in glioblastoma: therapeutic opportunities and challenges. Semin Oncol. 2011;38:243-253.
- Chi AS, Sorensen AG, Jain RK, Batchelor TT. Angiogenesis as a therapeutic target in malignant gliomas. Oncologist. 2009;14:621-636.
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-996.
- Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5 year analysis of the EORTC NCIC trial. Lancet Oncol. 2009;10:459-466.
- Clarke JL, Iwamoto FM, Sul J, et al. Randomized phase II trial of chemotherapy followed by either dose-dense or metronomic temozolomide for newly diagnosed glioblastoma. J Clin Oncol. 2009;27:3861-3867.
- Gállego Pérez-Larraya J, Ducray F, Chinot O, et al. Temozolomide in elderly patients with newly diagnosed glioblastoma and poor performance status: an ANOCEF phase II trial. J Clin Oncol. 2011;29:3050-3055.
- Vrendenburgh JJ, Desjardins A, Herndon JE 2nd, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol. 2007;25:4722-4729.
- Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733-4740.
- Raizer JJ, Grimm S, Chamberlain MC, et al. A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer. 2010;116:5297-5305.
- Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817-2823.
- Perry JR, Bélanger K, Mason WP, et al. Phase II trial of continuous dose-intense temozolomide in recurrent malignant glioma: RESCUE study. J Clin Oncol. 2010;28:2051-2057.
- Yung WK, Vredenburgh JJ, Cloughesy TF, et al. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro Oncol. 2010;12: 1061-1070.
- Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003.
- Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6:39-51.
- A Study of Avastin (Bevacizumab) in Combination With Temozolomide and Radiotherapy in Patients With Newly Diagnosed Glioblastoma. http://clinical trials.gov/ct2/show/NCT00943826. Accessed July 1, 2011.
- Radiation Therapy Oncology Group. RTOG 0825 protocol information. www.rtog.org/ClinicalTrials/ProtocolTable/StudyDetails.aspx?study=0825. Accessed July 1, 2011.
- Lai A, Filka E, McGibbon B, et al. Phase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme: interim analysis of safety and tolerability. Int J Radiat Oncol Biol Phys. 2008;71:1372-1380.
- Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro Oncol. 2003;5:79-88.
- Keime-Guibert F, Chinot O, Taillandier L, et al. Radiotherapy for glioblastoma in the elderly. N Engl J Med. 2007;356:1527-1535.
- Wong ET, Hess KR, Gleason MJ, et al. Outcomes and prognostic factors in recurrent glioma patients enrolled onto phase II clinical trials. J Clin Oncol. 1999; 17:2572-2578.
- Lamborn KR, Yung WK, Chang SM, et al. Progression-free survival: an important end point in evaluating therapy for recurrent high grade gliomas. Neuro Oncol. 2008;10:162-170.
- Ballman KV, Buckner JC, Brown PD, et al. The relationship between six-month progression-free survival and 12-month overall survival end points for phase II trials in patients with glioblastoma multiforme. Neuro Oncol. 2007;9:29-38.
- Chamberlain MC. Emerging clinical principles on the use of bevacizumab for the treatment of malignant gliomas. Cancer. 2010;116:3988-3999.
- Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clin Med Insights Oncol. 2011;5:117-129.
- Stark-Vance V. Bevacizumab and CPT-11 in the treatment of relapsed malignant glioma. Proc Soc Neuro-Oncol. 2005;7:369. Abstract 342.
- Prados MD, Lamborn K, Yung WK, et al. A phase 2 trial of irinotecan (CPT-11) in patients with recurrent malignant glioma: a North American Brain Tumor Consortium study. Neuro Oncol. 2006;8:189-193.
- Cloughesy TH, Filka E, Kuhn J, et al. Two studies evaluating irinotecan treatment for recurrent malignant glioma using an every-3-week regimen. Cancer. 2003; 97:2381-2386.
- Chamberlain MC. Salvage chemotherapy with CPT-11 for recurrent glioblastoma multiforme. J Neurooncol. 2002;56:183-188.
- Yung WK, Albright RE, Olson J, et al. A phase II study of temozolomide vs procarbazine in patients with glioblastoma multiforme at first relapse. Br J Cancer. 2000;83:588-593.
- Mellinghoff IK, Wang MY, Vivanco I, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012-2024.
- Sathornsumetee S, Desjardins A, Vredenburgh, et al. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro Oncol. 2010;12:1300-1310.
- Terasaki M, Shibui S, Narita Y, et al. Phase I trial of personalized peptide vaccine for patients positive for human leukocyte antigen-A24 with recurrent or progressive glioblastoma multiforme. J Clin Oncol. 2011;29:337-344.
- Phuphanich S, Supko JG, Carson KA, et al. Phase I clinical trial of bortezomib in adults with recurrent malignant glioma. J Neurooncol. 2010;100:95-103.