Biologic drugs play an important role in healthcare and represent $232 billion in global revenue.1 Biologic drugs represent 25% of the total global pharmaceutical market.1 The number of biologic drugs approved by the US Food and Drug Administration (FDA) continues to increase, with 12 biologic drugs included in the 46 new molecular entities approved by the FDA in 2017.2 In addition, 10 new biologics were approved by the FDA in 2018, 40% of which were for the treatment of cancer.3 Biologic drugs are an important therapeutic option for patients with cancer and are used for the treatment of malignancies as well as for supportive care management.
What Is a Biosimilar?
A biosimilar is a drug that is highly similar, but not necessarily identical, to a biologic drug (ie, the reference drug) and shows no clinically significant differences in safety, purity, or potency.4 Biosimilar development does not establish safety and efficacy, but rather shows biosimilarity to the reference drug.4 Unlike generic drugs, biosimilars are not an exact copy of their reference drug because of differences in the manufacturing process.5
In Europe, the first biosimilar was approved in 2006, and since then, 48 biosimilars have been approved there.6 In the United States, the first biosimilar was approved in 2015.7 As of July 31, 2019, a total of 23 biosimilars have been approved by the FDA in the United States, 7 of which are currently available on the market.8,9
By 2020, 9 patents for the top 20 biologic drugs are set to expire.10 Between 2013 and 2024, 8 biologic drugs used in oncology will have patents expiring. With these pending patent expirations, there has been an increase in the number of biosimilars studied for the treatment of cancer, with more than 250 ongoing clinical trials.11
Hurdles to biosimilar development in the United States include regulatory requirements, patent issues, insurance coverage, and physician and patient preferences.5 However, if these problems can be overcome, biosimilars have the potential to continue to provide important healthcare treatment options while minimizing the costs for patients and for insurance companies.
The Association for Accessible Medicines and its biosimilars council estimate that by 2025, 1.2 million patients in the United States could gain access to biologic drugs because, “as biosimilars become more widely available in the United States, they expand therapeutic options, enhancing the likelihood that patients will be able to begin treatment with biologic medicines.”12
Biologic drugs are large molecules that “can be composed of sugars, proteins, or nucleic acids or complex combinations of these substances, or may be living entities such as cells and tissues.”13 Biologic drugs differ from other medications; they are typically large, complex molecules, which are difficult to synthesize.14 In contrast, small molecules are substantially lower in weight and can be synthesized using predictable chemical reactions.
Biologic drugs are manufactured from natural sources, including bacteria or mammalian and animal cells. They are susceptible to variability between manufacturers, as well as variability in separate batches of the drug. Minor changes in molecular structure and immunogenicity can arise between a biosimilar and its reference drug.14
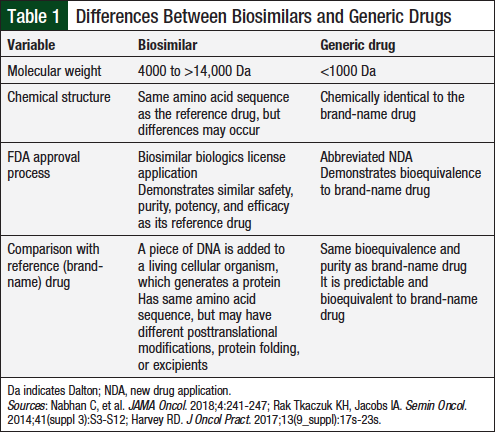
The terms “biosimilar” and “generic drug” should not be used interchangeably, because of the extensive differences between them.14 Biosimilars cannot be reproduced to the same degree as generic drugs, because of the inherent variability of biologic drugs.14 The Public Health Service (PHS) Act defines biosimilars as “a biologic product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components….[and] there are no clinically meaningful differences between the biologic product and reference product in terms of safety, purity, and potency of the product.”15 Table 1 illustrates the key differences between generic drugs and biosimilars.14,16,17
FDA Approval Process for Biosimilars
Biosimilars and generic drugs have different FDA approval processes. Abbreviated New Drug Application 505(j) is used to approve generic drugs.18 Preclinical and clinical data are typically not required to show safety and efficacy for generic drugs if the generic version performs similar to the original drug.18 The Biologics Price Competition and Innovation (BPCI) Act of 2009 was passed under the Affordable Care Act in 2010.19 As an amendment to the PHS Act, the BPCI Act establishes an “abbreviated licensure pathway for biological products shown to be biosimilar to or interchangeable with an FDA-licensed biological reference product.”15,19 Section 351(k) of the PHS Act describes requirements for the application of a proposed biosimilar drug and a proposed interchangeable drug.15,19
The FDA’s approach for evaluating biosimilarity is described as a “totality of the evidence,” because multiple studies are evaluated to determine similarity between a biosimilar and its reference drug.20 Information validating similarity is derived from analytical studies, animal studies, and clinical studies. Analytical studies are used to show biosimilarity, meaning that the biologic drug is “highly similar” to the reference drug. Animal studies are used to assess toxicities, whereas clinical trials, including pharmacokinetic, pharmacodynamics, and immunogenicity data, are used to establish potency, safety, and purity.20 A clinical trial comparing a biosimilar with its reference drug is also used to show that there are “no clinically meaningful differences between the biologic product and reference product.”20
There are other FDA requirements for the approval of biosimilars in addition to what is required for the approval of a reference drug.15 Biosimilars must utilize the same mechanism of action for the proposed condition(s) of use, and the condition(s) of use proposed in the labeling of the biosimilar must have been previously approved for the reference drug. Biosimilars need to have a similar route of administration, dosage, and strength, in addition to being developed with similar manufacturing processes as its reference drug, including processing, packaging, and storage.15
After a biosimilar has been approved by the FDA and is deemed to be similar to its reference drug, indications for the biosimilar may be extrapolated from the reference drug.21 Extrapolation is defined as “the approval of a biosimilar for use in an indication held by the reference product but not directly studied in a comparative clinical trial with the biosimilar.”21 The FDA bases extrapolation on sufficient scientific validation.
Several biosimilars are approved in the United States for all the same indications as their reference drug. Extrapolation is an important concept for the use of biosimilars in oncology, which eliminates the need for manufacturers to spend time and money on biosimilar research.
Biosimilar Naming
To ensure safe and proper use of biosimilars, the FDA released guidance pertaining to the nonproprietary naming of biologic drugs in January 2017.22 The proper name of a biologic drug, which is granted by the FDA in the license for use, often represents specific scientific characteristics of the drug. To prevent unintended substitution among biologics and biosimilars, the FDA requires that biologic drugs and biosimilars have the same nonproprietary name (ie, the name that is unprotected by trademark rights in the public domain). The FDA hopes to improve pharmacovigilance with this naming system, which will help track specific drugs.22
To assign a proper name to a drug, a core name or generic is designated by the United States Adopted Names Council for originator biologic drugs. This core name carries over if the biologic drug is a related biologic drug, a biosimilar, or an interchangeable drug. Four meaningless lowercase letters are then added after a hyphen to the end of the core name.22 For example, pegfilgrastim is the core name for one of several granulocyte colony-stimulating factors. Each of the biosimilars to pegfilgrastim contains “pegfilgrastim” as the core name, followed by a hyphen and 4 meaningless letters that are different in each biosimilar (for differentiation).
The FDA recognizes that this naming system may be problematic. This naming convention has the possibility to raise concerns among healthcare professionals because of the similarity between biologic drug names and biosimilar names. One example is the electronic health records, where suffixes are not always displayed or can be cut off as a result of space or character requirements. This would make it hard to differentiate whether a patient was receiving a biologic drug or a biosimilar drug.
Outpatient pharmacists often do not have access to hospital records, so if the entire drug name is not transmitted correctly, it would be difficult for the pharmacist to verify which medication the patient is receiving. In addition, healthcare providers may incorrectly assume that because a drug has the same core name, it is interchangeable with another drug with that core name.22 However, with continued use and an increase in the number of approved biosimilar agents, the use of this naming convention should become familiar to providers and pharmacists.
Biosimilar Labeling Recommendations
The FDA has provided guidance on information to be included in the package inserts of biosimilar drugs. Because the establishment of efficacy is not required for a biosimilar to be approved by the FDA, data indicating that biosimilarity may not be relevant to healthcare providers. According to the FDA, a biosimilar labeling should include a description of the clinical data that supported the safety and efficacy of the reference drug.23 Biosimilar labels must meet the content and format requirements of the Physician Labeling Rule and the Pregnancy and Lactation Labeling Rule. Additional recommendations include relevant data and information from the reference drug labeling, such as indications, dosing regimens, and adverse effects. Only indications for which the biosimilar was approved are to be included in its label, even if the reference drug is indicated for other uses.23
In July 2018, the FDA released a guidance document regarding the labeling of biosimilars.23 The guidance document notes that a biosimilar label should include clinical study information only when necessary to inform the healthcare provider about safety and efficacy, but also should include relevant data and information from the reference drug label, such as indication(s) and dosing regimen(s). The guidance also provides approaches to specific sections that should be included in a biosimilar labeling. It is important to note that the guidance does not include labeling for interchangeable drugs within this document.23
Interchangeability
According to the BPCI Act, a biosimilar that is expected to produce the same clinical result as the reference drug is considered interchangeable.24 A drug that is interchangeable can be substituted for the reference drug without prescriber approval.24 To be considered interchangeable with its reference drug, the biosimilar must go through further evaluation and testing.25 The biologic drug must be biosimilar to the reference drug and is expected to produce the same clinical result as the reference drug in any patient.25 In addition, the risks for diminished efficacy and for adverse events when switching from a reference drug to a biosimilar must not be greater than the risk for using the reference drug without switching if the drug is administered more than once.25
The FDA has not yet published official guidance on interchangeability, although a draft guidance is available.26 To gain interchangeability status, manufacturers must undergo rigorous testing of their biosimilar, which requires time and an investment of money into the approval process to show “that the biosimilar is expected to produce the same clinical result as the reference product in any given patient.”24
Although biosimilars to date are not interchangeable with their reference drug, biosimilars are not inherently inferior to their reference drug. To gain FDA approval, biosimilars are expected to produce the same response as their reference drug in terms of safety, purity, and potency. To date, biosimilars do not have interchangeability status, because there are no data to support patients switching from a reference drug to a biosimilar and then back to the reference drug. With thorough testing that has met FDA standards, patients and prescribers should be assured that the interchangeable drug will produce the same clinical outcome as the reference drug.
Prescribing Biosimilars
If a biosimilar is deemed interchangeable with its reference drug by the FDA, the healthcare provider who prescribes the reference drug does not need to be consulted to substitute the reference drug with a biosimilar. State pharmacy laws, not the FDA, dictate the substitution of a biosimilar. Once biologic drugs are designated interchangeable, some states may permit pharmacy-level substitution, which would allow a pharmacist to substitute an interchangeable biosimilar for its reference drug without consulting the prescriber.25 Some states require that the pharmacist inform the patient and/or the prescriber of the substitution or that they retain records of the substitution, which may be time-consuming or burdensome to some practitioners.27
The Purple Book
Biologic drugs, biosimilars, and interchangeable biologic drugs licensed by the FDA under the PHS Act are listed in the “Purple Book.”28 The Purple Book lists biologic drugs, and, if applicable, its biosimilar drug, as well as information on interchangeability for each drug. The Purple Book also provides information on any existing reference drug exclusivity protecting a reference biologic drug.
The information provided includes the date a biologic drug was licensed under 351(a) of the PHS Act, whether the FDA evaluated the biologic drug for reference drug exclusivity under section 351(k)(7) of the PHS Act, and whether a biologic drug licensed under section 351(k) has been determined by the FDA to be biosimilar to or interchangeable with its reference drug.28
Biosimilars in Oncology
Currently, 14 approved biosimilars are indicated for oncology-related treatment or supportive care.8 The first-ever biosimilar to be approved by the FDA was filgrastim-sndz (Zarxio), which was approved in 2015 as a biosimilar to the reference drug filgrastim (Neupogen). In July 2018, filgrastim-aafi (Nivestym) was approved.7 These 2 biosimilars are approved for all the same indications as their reference drug, filgrastim, including use in patients with cancer who are receiving myelosuppressive chemotherapy; patients with acute myeloid leukemia receiving induction or consolidation chemotherapy; patients with cancer undergoing bone marrow transplantation, patients undergoing autologous peripheral blood progenitor-cell collection and therapy; and patients with severe chronic neutropenia.29-31
Filgrastim-sndz is only available as a prefilled syringe,29 whereas filgrastim and filgrastim-aafi are supplied in prefilled syringes and in single-dose vials for subcutaneous use or intravenous infusion.30,31
Bevacizumab-awwb (Mvasi), the first biosimilar approved for the treatment of cancer, as opposed to supportive care, was approved in September 2017 for the treatment of metastatic colorectal cancer, nonsquamous non–small-cell lung cancer, glioblastoma, metastatic renal-cell carcinoma, and persistent, recurrent, or metastatic cervical cancer.32
Biosimilars are studied in one indication, and then the data are extrapolated for approval in other indications. Unlike its reference drug bevacizumab (Avastin), bevacizumab-awwb is not approved for ovarian cancer, because its manufacturer has orphan drug exclusivity protection for 2 ovarian cancer indications until 2021.33,34 Bevacizumab-awwb is expected to be approved for use in ovarian cancer once the orphan drug exclusivity status expires.33,34
As of July 31, 2019, there are 5 FDA-approved biosimilars for trastuzumab (Herceptin), which is indicated for the treatment of human epidermal growth factor receptor (HER)2-overexpressing breast cancer or for HER2-overexpressing metastatic gastric or gastroesophageal junction adenocarcinoma.7,35,36 These biosimilars include trastuzumab-dkst (Ogivri), trastuzumab-pkrb (Herzuma), trastuzumab-dttb (Ontruzant), trastuzumab-qyyp (Trazimera), and trastuzumab-anns (Kanjinti).7
Pegfilgrastim-jmdb (Fulphila) was approved by the FDA in June 2018 as the first biosimilar to pegfilgrastim (Neulasta).37 Pegfilgrastim-jmdb is indicated to decrease the likelihood of infection in patients with nonmyeloid malignancies who are receiving myeloablative chemotherapy.38 Unlike pegfilgrastim, pegfilgrastim-jmdb is not approved for hematopoietic subsyndrome of acute radiation syndrome.38,39 Pegfilgrastim-jmbd is not available in a formulation for use with an on-body injector. Both drugs are available for subcutaneous administration via a single-dose prefilled syringe.38,39
Epoetin alfa-epbx (Retacrit) was approved in May 2018 as the first erythropoietin-stimulating biosimilar.40 Like its reference drugs, epoetin alfa-epbx is indicated for the treatment of anemia that results from chronic kidney disease, from HIV treatment with zidovudine, or from the effects of concomitant myelosuppressive chemotherapy.40,41 It is also indicated for reduction of allogeneic red blood cell transfusions in patients undergoing elective, noncardiac, nonvascular surgery.40,41
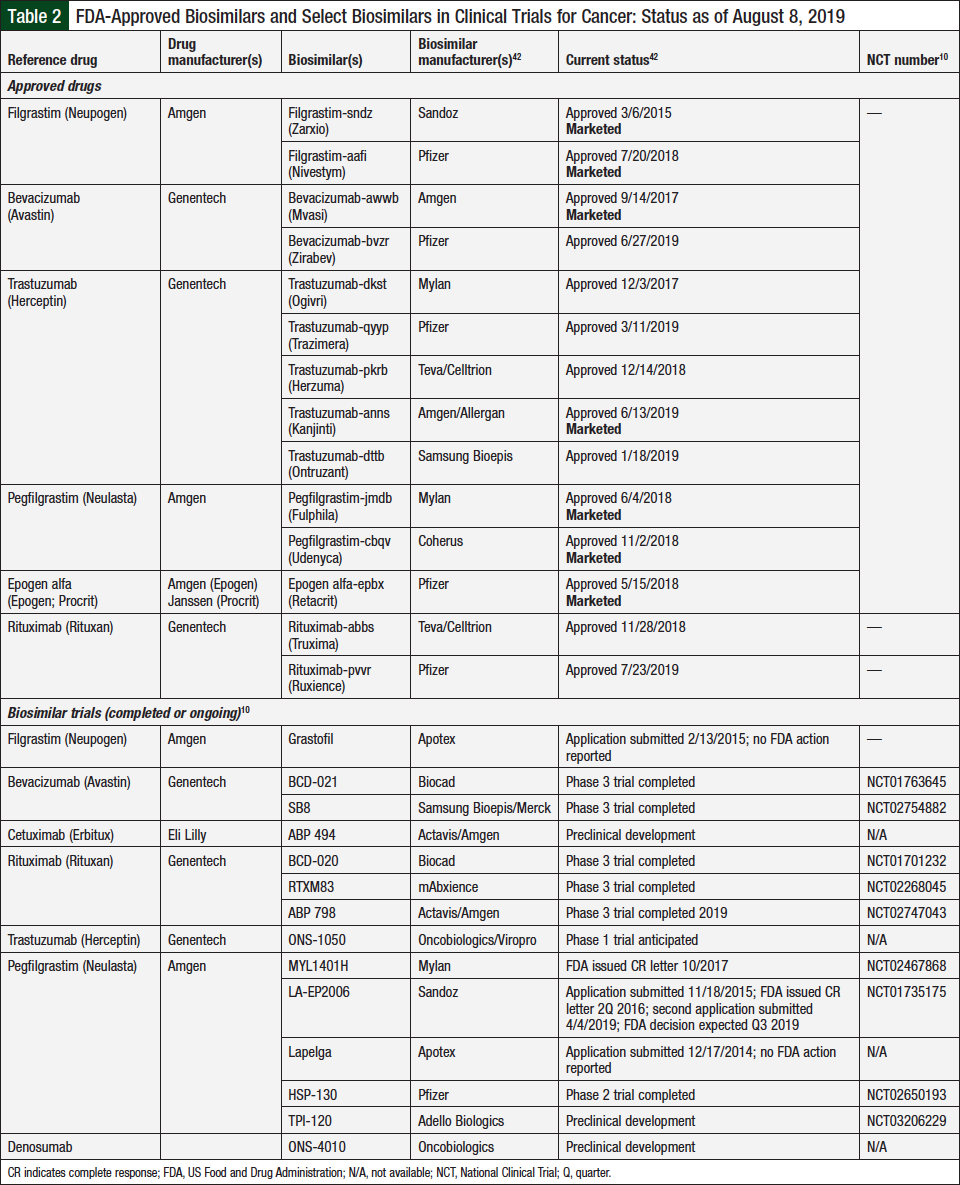
As noted, several biosimilars in oncology have been approved by the FDA, and more approvals are on the horizon. Table 2 lists the currently approved biosimilars in oncology and those in development by manufacturers that are actively seeking approval of their biosimilar for cancer treatment or for supportive care indications.10,42,43
Another important issue to consider is the use of biologic drugs with orphan drug status. An orphan drug is a medication that is used to treat rare diseases.44 Because few patients may be receiving the medication, orphan drug status gives a manufacturer incentive to develop and market a drug that may only be used for a small number of patients. This often results in high-cost medications. The impact that biosimilars, which carry a lower price, may have on the price of medications used for orphan drug indications remains unclear.44
Potential for Cost-Savings
The use of biosimilars may result in exponential cost-savings. A substantial increase in spending on prescription drugs in 2015 and 2016 in the United States resulted in large part from the increased use of specialty drugs, including biologic drugs.45 On average, biologic drugs are 20 times more expensive than traditional chemical drugs.46 Because of their high cost, biologic drugs alone contributed to 38% of prescription drug costs in 2015.45 The BPCI Act of 2009 was designed to initiate competition between manufacturers of biologic drugs.47
In 2017, the RAND Corporation issued a report entitled “Biosimilar Cost Savings in the United States.”45 In the report, Mulcahy and colleagues estimated “that biosimilars will lead to a reduction of $54 billion in direct spending on biologic drugs from 2017 to 2026, or about 3 percent of total estimated biologic spending over the same period, with a range of $24 to $150 billion.”45 Through their literature review, Mulcahy and colleagues noted that the key drivers of cost-savings include the number and timing of biosimilars in development and marketing, the acceptability of use among patients and prescribers, the cost of biosimilar development, changes in the US market size, payer coverage, cost-sharing, interchangeability, and other regulatory policies.45
Although these numbers sound promising, the anticipated cost-savings with the use of biosimilars are predicted to be modest. Worldwide, the cost of biosimilars is approximately 20% to 30% less than that of reference drugs.48 The cost-savings will depend on multiple factors, including the cost of the reference drug, its sales, and the degree of competition.49
Challenges of Integrating Biosimilars into Practice
The use of biosimilars in patients with cancer presents an opportunity to educate prescribers and patients. According to a survey of 376 US oncologists, prescribers lacked knowledge and understanding of the effects of biologic drugs and biosimilars that share the same nonproprietary name.50 Confusion also arose regarding whether biologic drugs and biosimilars are structurally and therapeutically identical.50
The American Society of Clinical Oncology (ASCO) released a statement in 2018 emphasizing the need for postmarketing evidence to enhance physician and patient confidence in the use of biosimilars.51 The FDA approval process for biosimilars is shorter than that for biologic drugs because the approval of biosimilars relies heavily on structural, functional, and pharmacologic data compared with the efficacy and safety data that are necessary for the approval of original biologic drugs. ASCO suggests the use of its CancerLinQ database, which provides deidentified patient data, as well as the Sentinel Initiative—the pending FDA surveillance system that is designed to monitor safety issues in clinical trials—to gather more data on the use of biosimilars.51
Patients may become concerned if they are switched to a biosimilar for treatment if their disease has been in remission or is controlled. A survey of 1181 patients conducted by the European Federation of Crohn’s and Ulcerative Colitis Associations showed that only 38% of people had heard of biosimilars.52 The most common concerns about biosimilars among respondents were safety and efficacy. Approximately 56% of respondents thought that the cost-savings of biosimilars should not come before safety and efficacy.52 When it comes to interchangeability, approximately 21% of respondents would be against the idea if the patient was not made aware of the switch. Only approximately 27% of patients would trust their physician and less than 1% would trust their pharmacist to make the decision to include a biosimilar in their treatment plan.52 As pharmacists, it is imperative to advise prescribers and patients of the safety and efficacy of biosimilars.
The potential for immunogenicity is another concern with biosimilars. Biologic drugs are inherently immunogenic because of their size and complexity, and this potential exists between different batches of a reference biologic drug.53 In immune-mediated reactions, the body recognizes that the biologic drug and biosimilar are foreign, and, in turn, produces antibodies, which leads to decreased efficacy and/or increased side effects.53 Although it is possible for biosimilars to cause similar reactions, they are not associated with an increase in the number or severity of reactions. As part of the PIONEER clinical trial, which was a phase 3 clinical trial in patients with breast cancer that led to the FDA approval of filgrastim-sndz, patients alternated back and forth between the biosimilar and the reference drug throughout 6 chemotherapy cycles. There were no differences in safety or efficacy among the drugs, and antibodies were not detected.53 This is one way interchangeability can be demonstrated, but there remains a need for principles to guide interchangeability to address the safety and efficacy of alternating between the reference drug and the biosimilar.
Filgrastim-sndz and pegfilgrastim-jmdb are available only as prefilled syringes.29,40 In patients who weigh less than 45 kg and are prescribed a dose of filgrastim of <300 μg (0.5 mL) or a dose of pegfilgrastim of <6 mg (0.6 mL), it is challenging to measure an accurate dose when the medication is provided to the patient in a prefilled syringe.31,39 Certain drugs and biosimilars are available in a prefilled syringe that does not have graduation marks to obtain the correct weight-based dose, and the direct administration of these drugs is not recommended in patients who weigh less than 45 kg.25,38,39 The prescribing information for some of these drugs contains a table to guide providers in dosing pediatric patients who weigh less than 45 kg.41,44
The European Medicines Agency, which is the governing body of biosimilar approval in Europe, does not mention interchangeability, but rather allows each country to establish individually if a biosimilar is interchangeable with its reference drug.54 In the United States, none of the available biosimilars for oncology treatment or supportive care are designated as interchangeable. Extensive studies are required to show interchangeability in the United States. Because of confidentiality issues, the FDA cannot disclose which applications have been submitted to gain interchangeability status. However, now that the FDA has released its draft guidance on biosimilar interchangeability, interchangeable biosimilars are expected to be available soon.55
In September 2017, the FDA issued its “Statistical Approaches to Evaluate Analytical Similarity,” which provided advice on the evaluation of analytical similarity to sponsors interested in developing biosimilars.56 This draft guidance described “the type of information a sponsor of a proposed biosimilar product should obtain about the structural/physicochemical and functional attributes of the reference product, how that information is used in the development of an analytical similarity assessment plan for the proposed biosimilar, and the statistical approaches recommended for evaluating analytical similarity.”56
In June 2018, the FDA withdrew its draft guidance after the public raised a wide range of issues regarding the impact of cost and efficacy of biosimilar development.57 The FDA stated that future guidance will include state-of-the-art techniques to evaluate data that show that a biosimilar is highly similar to its reference drug. The goal will be to address the potential challenges that biosimilar manufacturers may face, including methods to analyze data to account for the potential variability of reference drugs with different lot numbers and development procedures that are efficient but that do not compromise scientific standards.57
According to the Centers for Medicare & Medicaid Services (CMS), Medicare Part B will no longer group biosimilars and reference drugs in the same billing code for dates of service on or after April 1, 2018.58 Before this, Medicare Part B paid for biosimilars under the Physician Fee Schedule based on the average sales price (ASP), and biosimilars and reference drugs were grouped together.58 This meant physicians were reimbursed the same amount for all biosimilars of the same reference drug, regardless of the biosimilar that was administered.59
After the update by CMS, each biosimilar and its reference drug will have an individualized procedure code,58 which is also known as the Healthcare Common Procedure Coding System (HCPCS).59 Previously, biosimilars and their referenced drugs shared the same HCPCS, so physicians were reimbursed the same amount regardless of which drug they prescribed.59 Going forward, physicians will be reimbursed for the ASP of the biosimilar plus 6% of the reference drug’s ASP.60 Therefore, providers will get a small incentive to administer a biosimilar.
The Bipartisan Budget Act of 2018 outlines the changes to prescription drug coverage under Medicare Part D.61 Beginning in 2019, patient coinsurance was reduced from 30% to 25% of the reference drug, which will expedite closure of the coverage gap.60 As part of the Bipartisan Budget Act of 2018, biosimilars were included in the coverage gap discount; before this, biosimilars were not eligible for the coverage gap discount, and patients would have had to pay 50% to 70% more for the biosimilar.60 As a result, drug manufacturers may be deterred from marketing biosimilars in the future, because they previously had no obligation to offer a discount on generic drugs.60
An additional concern relates to automatic substitution. Some state laws dictate that pharmacists have the ability to automatically substitute a biosimilar for a reference drug if the biosimilar is deemed interchangeable.62 Some stakeholders believe that state substitution laws will require revisions to include biosimilars, which may vary.62 The FDA’s “interchangeable” designation may be sufficient in some states, but others may require additional information. This may affect prescribing, as physicians would have to order the specific drug.63 Furthermore, as of July 31, 2019, a total of 23 biosimilars have been approved in the United States, 14 of which carry indications for use in oncology, but so far only 7 of these are available on the market.42
Conclusion
The use of biosimilars in patients with cancer in the United States is rapidly evolving. As patents for biologic drugs expire, manufacturers are submitting applications to the FDA for the approval of biosimilars. The FDA has issued either guidance or draft guidance on naming recommendations, labeling, and the interchangeability of biosimilars. Of note, among the 23 biosimilars approved in the United States by the end of July 2019, 14 carry indications for use in oncology. Nevertheless, of these 14 biosimilars, only 7 (50%) are currently available on the market. The other 7 approved biosimilars are yet to be released to the market.
The potential cost-savings to patients and insurance companies make biosimilars an ideal treatment option. Any concerns regarding the use of biosimilars arise from confusion regarding similarities and differences between biosimilars and their reference drugs, immunogenicity, patient reluctance, and dosing in pediatric patients. However, all these challenges are opportunities for the evolution of biosimilars.
Author Disclosure Statement
Dr Pittman, Dr Wern, and Dr Glode have no conflicts of interest to report.
References
- Kent D, Rickwood S, Di Biase S. Disruption and maturity: the next phase of biologics. QuintilesIMS. www.iqvia.com/-/media/iqvia/pdfs/nemea/uk/disruption_and_maturity_the_next_phase_of_biologics.pdf. Accessed August 7, 2019.
- de la Torre BG, Albericio F. The pharmaceutical industry in 2017. An analysis of FDA drug approvals from the perspective of molecules. Molecules. 2018;23:533.
- US Food and Drug Administration. Novel Drug Approvals for 2018. September 27, 2018. www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/novel-drug-approvals-2018. Accessed August 7, 2019.
- US Food and Drug Administration. Biosimilar development, review, and approval. October 23, 2017. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580429.htm. Accessed July 21, 2018.
- Zelenetz AD, Ahmed I, Braud EL, et al. NCCN biosimilars white paper: regulatory, scientific, and patient safety perspectives. J Natl Compr Canc Netw. 2011;9(suppl 4):S1-S22.
- Harston A. How the U.S. Compares to Europe on Biosimilar Approvals and Products In the Pipeline. Rothwell Figg IP Professionals: Biologics & Biosimilars. https://www.biosimilarsip.com/2019/05/07/how-the-u-s-compares-to-europe-on-biosimilar-approvals-and-products-in-the-pipeline-4/. Accessed August 7, 2019.
- US Food and Drug Administration. Biosimilar product information. July 20, 2018. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580432.htm. Accessed July 21, 2018.
- Davio K. Amgen and Allergan launch Mvasi and Kanjinti, the first anticancer biosimilars, in the United States. July 19, 2019. www.centerforbiosimilars.com/news/amgen-and-allergan-launch-mvasi-and-kanjinti-the-first-anticancer-biosimilars-in-the-united-states. Accessed July 31, 2019.
- US Food and Drug Administration. Biosimilar product information. www.fda.gov/drugs/biosimilars/biosimilar-product-information. Accessed July 18, 2019.
- Cohen J. What’s holding back market uptake of biosimilars? June 20, 2018. www.forbes.com/sites/joshuacohen/2018/06/20/whats-holding-back-market-uptake-of-biosimilars/#34cbc0e2691a. Accessed August 7, 2019.
- ClinicalTrials.gov. Search results: biosimilar + oncology. https://clinicaltrials.gov/ct2/results?cond=oncology&term=biosimilar&cntry=&state=&city=&dist=. Accessed August 7, 2019.
- AAM Biosimilars Council releases 2017 report on patient access to biosimilars in the United States. September 12, 2017. www.prnewswire.com/news-releases/aam-biosimilars-council-releases-2017-report-on-patient-access-to-biosimilars-in-the-united-states-300517960.html. Accessed November 19, 2018.
- US Food and Drug Administration. What are “biologics” questions and answers. February 6, 2018. www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/CBER/ucm133077.htm. Accessed July 21, 2018.
- Nabhan C, Parsad S, Mato AR, Feinberg BA. Biosimilars in oncology in the United States: a review. JAMA Oncol. 2018;4:241-247.
- US Food and Drug Administration. Public Health Service Act. 2018. https://legcounsel.house.gov/Comps/PHSA-merged.pdf. Accessed July 21, 2018.
- Rak Tkaczuk KH, Jacobs IA. Biosimilars in oncology: from development to clinical practice. Semin Oncol. 2014;41(suppl 3):S3-S12.
- Harvey RD. Science of biosimilars. J Oncol Pract. 2017;13(9_suppl):17s-23s.
- US Food and Drug Administration. Determining whether to submit an ANDA or a 505(b)(2) application: guidance for industry. May 2019. www.fda.gov/media/124848/download. Accessed July 19, 2019.
- US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry. April 2015. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed July 21, 2018.
- US Food and Drug Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product: guidance for industry. December 2016. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed November 16, 2017.
- Curigliano G, O’Connor DP, Rosenberg JA, Jacobs I. Biosimilars: extrapolation for oncology. Crit Rev Oncol Hematol. 2016;104:131-137.
- US Food and Drug Administration. Nonproprietary naming of biological products: guidance for industry. January 2017. www.fda.gov/downloads/drugs/guidances/ucm459987.pdf. Accessed November 16, 2017.
- US Food and Drug Administration. Labeling for biosimilar products: guidance for industry. July 2018. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM493439.pdf. Accessed July 21, 2018.
- Oskouei ST. Biosimilar interchangeability: 9 things to consider. April 2, 2018. www.centerforbiosimilars.com/contributor/sonia-oskouei/04/2018/biosimilar-interchangeability-9-things-to-consider. Accessed July 21, 2018.
- US Food and Drug Administration. Prescribing biosimilar and interchangeable products. October 23, 2017. www.fda.gov/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/ucm580430.htm. Accessed July 21, 2018.
- US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product: guidance for industry. May 2019. www.fda.gov/media/124907/download. Accessed July 22, 2019.
- Cauchi R. State laws and legislation related to biologic medications and substitutions of biosimilars. October 22, 2018. www.ncsl.org/research/health/state-laws-and-legislation-related-to-biologic-medications-and-substitution-of-biosimilars.aspx. Accessed November 19, 2018.
- Lal R. The Purple Book. FDA/CDER SBIA Chronicles. November 18, 2014. www.fda.gov/downloads/drugs/developmentapprovalprocess/smallbusinessassistance/ucm423462.pdf. Accessed July 21, 2018.
- Zarxio (filgrastim-sndz) injection, for subcutaneous or intravenous use [prescribing information]. Princeton, NJ: Sandoz; April 2018.
- Nivestym (filgrastim-aafi) injection, for subcutaneous or intravenous use [prescribing information]. Lake Forest, IL: Pfizer; July 2018.
- Neupogen (filgrastim) injection, for subcutaneous or intravenous use [prescribing information]. Thousand Oaks, CA: Amgen; June 2018.
- Mvasi (bevacizumab-awwb) injection, for intravenous use [prescribing information]. Thousand Oaks, CA: Amgen; June 2019.
- Taylor P. Amgen’s Mvasi is first FDA-approved cancer biosimilar. September 15, 2017. www.pmlive.com/pharma_news/amgens_mvasi_is_first_fda-approved_cancer_biosimilar_1205426. Accessed July 21, 2018.
- Avastin (bevacizumab) injection, for intravenous use [prescribing information]. South San Francisco, CA: Genentech; June 2019.
- Ogivri (trastuzumab-dkst) for injection, for intravenous use [prescribing information]. Steinhausen, Switzerland: Mylan; April 2019.
- Herceptin (trastuzumab) for injection, for intravenous use [prescribing information]. South San Francisco, CA: Genentech; November 2018.
- US Food and Drug Administration. FDA approves first biosimilar to Neulasta to help reduce the risk of infection during cancer treatment. June 4, 2018. www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm609805.htm. Accessed July 21, 2018.
- Fulphila (pegfilgrastim-jmdb) injection, for subcutaneous use [prescribing information]. Steinhausen, Switzerland: Mylan; May 2019.
- Neulasta (pegfilgrastim) injection, for subcutaneous use [prescribing information]. Thousand Oaks, CA: Amgen; April 2019.
- Retacrit (epoetin alfa-epbx) injection, for intravenous or subcutaneous use [prescribing information]. Lake Forest, IL: Pfizer; January 2019.
- Procrit (epoetin alfa) injection, for intravenous or subcutaneous use [prescribing information]. Horsham, PA: Janssen; July 2018.
- SM Health Communications. Biosimilar approval status. Biosimilars Review & Report. Updated July 24, 2019. https://biosimilarsrr.com/us-biosimilar-filings/. Accessed July 22, 2018.
- Lyman GH, Zon R, Harvey RD, Schilsky RL. Rationale, opportunities, and reality of biosimilar medications. N Engl J Med. 2018;378:2036-2044.
- Davio K. Could biosimilars of orphan drugs reduce patients’ cost burdens? October 30, 2018. www.centerforbiosimilars.com/news/could-biosimilars-of-orphan-drugs-reduce-patients-cost-burdens. Accessed November 19, 2018.
- Mulcahy AW, Hlávka JP, Case SR. Biosimilar cost savings in the United States: initial experience and future potential. 2017. www.rand.org/content/dam/rand/pubs/perspectives/PE200/PE264/RAND_PE264.pdf. Accessed July 21, 2018.
- Fuhr JP. Biosimilars can save lives and cost less. August 8, 2014. www.forbes.com/sites/realspin/2014/08/08/biosimilars-can-save-lives-and-cost-less/#45b8a5592af3. Accessed July 21, 2018.
- US Food and Drug Administration. Implementation of the Biologics Price Competition and Innovation Act of 2009. February 12, 2016. www.fda.gov/drugs/guidance-compliance-regulatory-information/implementation-biologics-price-competition-and-innovation-act-2009. Accessed August 8, 2019.
- Singh SC, Bagnato KM. The economic implications of biosimilars. Am J Manag Care. 2015;21(16 suppl):S331-S340.
- Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol. 2017;3:596-610.
- Dolan C. Opportunities and challenges in biosimilar uptake in oncology. Am J Manag Care. 2018;24(11 suppl):S237-S243.
- Lyman GH, Balaban E, Diaz M, et al. American Society of Clinical Oncology statement: biosimilars in oncology. J Clin Oncol. 2018;36:1260-1265.
- Peyrin-Biroulet L, Lönnfors S, Roblin X, et al. Patient perspectives on biosimilars: a survey by the European Federation of Crohn’s and Ulcerative Colitis Associations. J Crohns Colitis. 2017;11:128-133.
- Rifkin RM, Peck SR. Biosimilars: implications for clinical practice. J Oncol Pract. 2017;13(9_suppl):24s-31s.
- European Medicines Agency. Biosimilars in the EU: information guide for healthcare professionals. 2017. www.ema.europa.eu/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf. Accessed November 19, 2018.
- Brennan Z. FDA: interchangeable biosimilar approvals expected within 2 years. June 26, 2017. www.raps.org/Regulatory-Focus/News/2017/06/26/27969/FDA-Interchangeable-Biosimilar-Approvals-Expected-Within-2-Years/. Accessed July 21, 2018.
- US Food and Drug Administration. Statistical approaches to evaluate analytical similarity; draft guidance for industry; availability. Fed Regist. 2017;82:44425-44426.
- US Food and Drug Administration. FDA withdraws draft guidance for industry: statistical approaches to evaluate analytical similarity. June 21, 2018. www.fda.gov/Drugs/DrugSafety/ucm611398.htm. Accessed July 21, 2018.
- Centers for Medicare & Medicaid Services. Part B biosimilar biological product payment and required modifiers. February 2, 2018. www.cms.gov/Medicare/Medicare-Fee-for-Service-Part-B-Drugs/McrPartBDrugAvgSalesPrice/Part-B-Biosimilar-Biological-Product-Payment.html. Accessed July 21, 2018.
- Syrop J. CMS reverses its policy on biosimilar reimbursement, will issue unique J-codes. November 3, 2017. www.centerforbiosimilars.com/news/cms-reverses-its-policy-on-biosimilars-reimbursement-will-issue-unique-jcodes. Accessed July 21, 2018.
- Chen M. Medicare biosimilar reimbursement: hopes for cost savings, a dream deferred. April 18, 2018. www.managedcaremag.com/pharmdcorner/medicare-biosimilar-reimbursement-hopes-cost-savings-dream-deferred. Accessed July 28, 2018.
- Bipartisan Budget Act of 2018, HR 1892, 115th Cong (2018). www.congress.gov/115/plaws/publ123/PLAW-115publ123.pdf. Accessed July 24, 2019.
- American Cancer Society Cancer Action Network. Understanding biologic medicines from the cancer patient perspective. January 2013. http://action.fightcancer.org/site/DocServer/ACSCAN-Biosimilars-Primer.pdf?docID=22449.%C2%A0. Accessed July 21, 2018.
- Hirsch BR, Lyman GH. Biosimilars: a cure to the U.S. health care cost conundrum? Blood Rev. 2014;28:263-268.