The discovery of predictive biomarkers in the development of targeted therapies has revolutionized cancer treatment. A predictive biomarker is a measure characteristic that provides information on the probability of response to specific therapies. Predictive biomarkers include various germline or somatic mutations, gene rearrangements, proteins, and other molecules, and may be detected through a US Food and Drug Administration (FDA)-approved companion diagnostic test or another method. Using predictive biomarkers to guide therapeutic decisions is necessary to optimize patient outcomes; predictive biomarkers are an essential component of clinical practice guidelines.1,2
By contrast, prognostic biomarkers influence patient survival or disease recurrence outcomes, regardless of any therapeutic intervention. Some prognostic biomarkers are also predictive biomarkers, but this review is focused exclusively on predictive biomarkers.
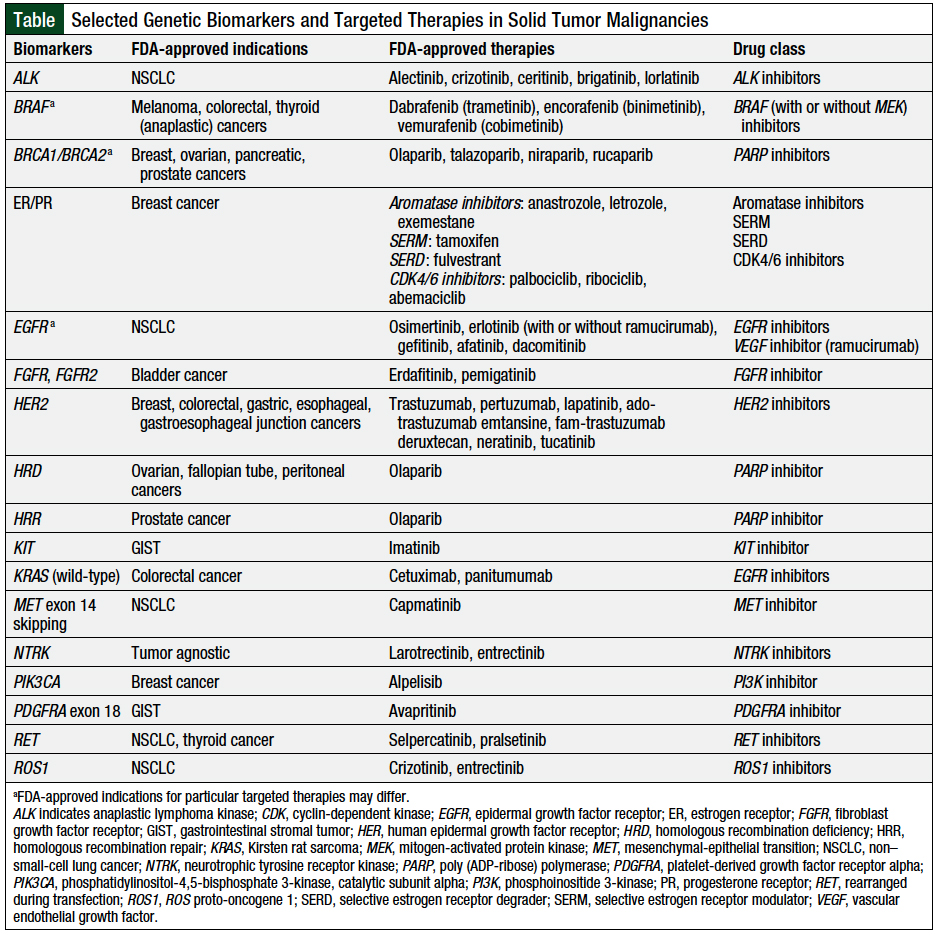
The goal of this review article is to describe the current FDA-approved targeted therapies and their indications in solid tumor malignancies through December 1, 2020, based on predictive biomarkers (Table).
ALK Inhibitors
Anaplastic lymphoma kinase (ALK) gene alterations were first discovered in anaplastic large-cell lymphoma.3 ALK gene rearrangements, also known as ALK fusions, are usually an inversion on chromosome 2 that creates ALK oncogenic fusion proteins, which lead to increased tumor proliferation.4 In 2007, ALK alterations were discovered for the first time in a solid tumor malignancy, namely, non–small-cell lung cancer (NSCLC), which led to significant advances in the treatment of NSCLC in the metastatic setting.4
Alectinib, crizotinib, ceritinib, brigatinib, and lorlatinib all inhibit the phosphorylation of ALK, which decreases tumor-cell growth in ALK-positive tumors.3 Crizotinib, a first-generation ALK inhibitor, has poor central nervous system penetration compared with later-generation ALK inhibitors. All the ALK inhibitors may be used for the treatment of ALK-positive metastatic NSCLC; ALK mutations occur in 3% to 5% of patients with NSCLC and are usually associated with being a nonsmoker and adenocarcinoma histology.3
The side effects associated with ALK inhibitors include bradycardia, gastrointestinal (GI) toxicity, visual disturbances, and pulmonary adverse events.5
ALK tyrosine inhibitors have been shown to improve overall survival, progression-free survival, and quality of life in patients with metastatic NSCLC.6-8
BRAF Inhibitors
The mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) pathway plays an essential role in cell proliferation.9 Alterations in the MAPK/ERK pathway, such as activating mutations in receptor tyrosine kinase (RTK) or mutations in the kinase rapidly accelerating fibrosarcoma (RAF), can lead to cancer-cell growth.9
BRAF is a kinase that activates the MAPK/ERK-signaling pathway. Mutations in BRAF can lead to uncontrolled cell proliferation, which is seen in approximately 40% to 60% of patients with cutaneous melanoma.10 The most common BRAF mutations are BRAF V600E (80%) and BRAF V600K (10%-30%).10,11
BRAF inhibitors (ie, dabrafenib, encorafenib, and vemurafenib) are frequently used in combination with mitogen-activated protein kinase (MEK) inhibitors (ie, trametinib, binimetinib, and cobimetinib). MEK is another protein downstream of BRAF, and the inhibition of these 2 targets has been shown to improve overall survival and progression-free survival compared with monotherapy BRAF inhibition; for example, the use of dabrafenib monotherapy versus dabrafenib plus trametinib; encorafenib monotherapy versus encorafenib plus binimetinib12; and vemurafenib monotherapy versus vemurafenib plus cobimetinib.13
The combination of dabrafenib and trametinib is FDA-approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutation, as well as for the treatment of metastatic NSCLC with BRAF V600E mutation or locally advanced or metastatic anaplastic thyroid cancer with BRAF V600E mutation.
Encorafenib plus binimetinib is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutation; encorafenib is also approved in combination with cetuximab for the treatment of metastatic colorectal cancer (CRC) with BRAF V600E mutation.
Patients with metastatic CRC and BRAF V600E mutation are unlikely to respond to the epidermal growth factor receptor (EGFR) cetuximab without the addition of a BRAF inhibitor.14 BRAF is downstream of EGFR; thus, the lack of BRAF inhibition bypasses EGFR inhibition through the constitutive activation of BRAF mutation, which leads to continued tumor growth.14 By contrast, the combination of cobimetinib plus vemurafenib is approved for the treatment of unresectable or metastatic melanoma with BRAF V600E or BRAF V600K mutation.
The drug class–related side effects of BRAF inhibitors, with or without MEK inhibitors, include dermatologic adverse events, fevers, secondary cutaneous malignancies, hemorrhage, cardiomyopathy, and ocular toxicities.15
Other than cutaneous melanoma, BRAF mutations also occur in anaplastic thyroid cancer (20%-50%), colon cancer (5%-15%), and NSCLC (3.5%-4%), among other tumor types.16-18
BRCA1 and BRCA2 Mutations
BRCA1 and BRCA2 are tumor-suppressor genes that help to repair breaks in DNA. Mutations in these genes lead to uncontrolled cell proliferation. BRCA1 and BRCA2 mutations can be germline or somatic, and poly (ADP-ribose) polymerase (PARP) inhibitors have shown efficacy in treating various germline BRCA1 and BRCA2 mutation–positive solid-tumor malignancies.19
PARP enzymes are involved in DNA repair and DNA transcription. PARP inhibitors are particularly active in tumors with BRCA1 or BRCA2 mutations, because these tumors lack the ability to repair DNA strand breaks as a result of a deficiency in homologous recombination repair (HRR).20
PARP inhibitors are approved for the treatment of ovarian, breast, pancreatic, and prostate cancers. The incidence rates of BRCA1 or BRCA2 mutations in these tumor types are 5% to 15%, approximately 5%, 4% to 7%, and 0.9% to 5.3%, respectively.20-25 The PARP inhibitors class includes olaparib, talazoparib, niraparib, and rucaparib, and the PARP inhibitor class side effects include bone marrow suppression, fatigue, rash, GI toxicities, and secondary malignancies.26
Estrogen and Progesterone Receptors
Hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative breast cancer is the most common (87%) female breast cancer subtype.27 Patients are considered to have HR-positive disease if estrogen receptor (ER) or progesterone receptor (PR) is expressed in 1% to 100% of the cells evaluated.28
The use of endocrine therapies is the standard of care for the treatment or prevention of recurrent HR-positive breast cancer because these therapies have shown to improve overall survival or disease-free survival. Limited data are available to support the benefits of endocrine therapy in ER-low–positive (1%-10% expression) and ER-negative, PR-positive breast cancer than in ER-positive (>10% expression) breast cancer.28
The use of a specific endocrine therapy depends on several factors, including patient age, menopause status, tolerability, and survival benefit.29 Patients may be receiving selective ER modulators, such as tamoxifen and/or aromatase inhibitors, for up to 10 years. Tamoxifen inhibits ERs on breast cancer tumor cells, resulting in decreased cell proliferation.29 By contrast, aromatase inhibitors, such as letrozole, anastrozole, or exemestane, ultimately prevent the generation of estrogens.30 Selective ER modulators and aromatase inhibitors may lead to hot flashes, mood changes, bone mineral density changes, and cardiovascular complications.29
Patients with ER- or PR-positive and HER2-negative breast cancer may be candidates for treatment with cyclin-dependent kinase (CDK)4/6 inhibitors. CDK4/6 inhibitors block CDK4 and CDK6 from binding to cyclin D1 and subsequently preventing retinoblastoma protein phosphorylation and gene transcription.31
Cyclin D1 is more frequently overexpressed and amplified in ER-positive breast cancer compared with ER-negative breast cancer.32 It has been shown that ER-positive breast cancer, regardless of HER2 status, is the most sensitive breast cancer subtype to CDK4/6 inhibition.31,32
The use of CDK4/6 inhibitors (ie, palbociclib, ribociclib, or abemaciclib) with endocrine therapy is currently the preferred first-line regimen in patients with advanced or metastatic HR-positive, HER2-negative breast cancer, because this combination has been shown to improve overall survival.28,33-35
The drug-class adverse events for CDK4/6 include bone marrow suppression, GI toxicities, corrected QT prolongation (ie, ribociclib), and rarely pulmonary side effects.36
EGFR Inhibitors
EGFR is an RTK located on the epithelial cells and is amplified in a variety of malignancies. EGFR mutations lead to constitutive activation and cancer growth. Therefore, multiple EGFR inhibitors have been developed to hinder this signaling process. Approximately 50% of Asian patients and 10% of Caucasian patients with NSCLC have disease associated with EGFR mutations.37 Treatment with oral EGFR inhibitors, including erlotinib, gefitinib, afatinib, dacomitinib, and osimertinib, in patients with EGFR-positive NSCLC has been shown to improve overall survival and/or progression-free survival.38
Specific EGFR mutations, such as exon 20 insertions and T790M, lead to resistance to most EGFR inhibitors, except osimertinib, which has been shown effective in these patients.6,38,39 In addition, osimertinib and dacomitinib are FDA-approved for the treatment of patients with metastatic NSCLC and EGFR exon 19 deletion or exon 21 L858R substitution mutation.
The drug-class adverse events include GI side effects, cardiotoxicity, rashes, ocular side effects, and pulmonary side effects.40
FGFR Inhibitors
Fibroblast growth factor receptor (FGFR) is a transmembrane and is part of the RTK family. When the kinase is bound by its ligand, fibroblast growth factor, the downstream activation of RAS/MAPK and phosphatidylinositol 3-kinase (PI3K)/Akt pathways occur.41
Genetic alterations and dysregulation in FGFR can lead to tumor-cell proliferation.41
Several mechanisms may cause excessive FGFR signaling, including upregulated FGFR or FGFR expression, mutations, or chromosomal rearrangements in the genes encoding FGFR, impaired termination of FGFR signaling, or defective degradation of FGFR.41
FGFR genetic alterations occur in approximately 21% of patients with bladder cancer and in approximately 15% to 20% of patients with cholangiocarcinoma.42-44
Two FGFR inhibitors were approved by the FDA by December 1, 2020. Erdafitinib is a pan-FGFR1-4 tyrosine kinase inhibitor that is approved for the treatment of urothelial carcinoma with FGFR2 or FGFR3 gene alterations, whereas pemigatinib is an FGFR kinase inhibitor approved for the treatment of cholangiocarcinoma that expresses FGFR2 fusions or rearrangements.
The adverse events associated with FGFR inhibitors include hyperphosphatemia and ocular adverse events.45
Homologous Recombination Deficiency/Homologous Recombination Repair
Patients with homologous recombination deficiency (HRD)-positive status (which involves a deleterious or suspected deleterious BRCA mutation or genomic instability) have a decreased ability to conduct normal DNA repair, which makes the tumor more susceptible to PARP inhibition.46 PARP inhibitors, such as olaparib and niraparib, are also effective (in addition to targeting BRCA mutations) for the treatment of patients with genomic instability. Therefore, patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer and HRD-positive status are eligible for treatment with olaparib plus bevacizumab or with niraparib monotherapy.46,47
Based on the PAOLA-1 clinical trial evaluating olaparib, HRD scores of at least 42 are considered positive results.46 Olaparib is FDA approved for the treatment of metastatic castration-resistant prostate cancer in patients with germline or somatic HRR gene mutations after receiving enzalutamide or abiraterone treatment, based on the results of the PROfound clinical trial.48
The 15 HRR gene mutations in the PROfound clinical trial included ATM, BRCA1, BRCA2, BARD1, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L. Gene mutations that affect DNA damage repair, such as those listed above, can occur in up to 30% of patients with prostate cancer.48 HRR gene mutations are involved in DNA repair, and PARP inhibitors work synergistically, by disrupting tumor-cell repair.48
HER2 Inhibitors
HER2 is a transmembrane RTK that belongs to the EGFR family and functions by stimulating growth factor–signaling pathways, such as the PI3K/Akt pathway.49 The overexpression of HER2 leads to the constitutive activation of this pathway and subsequent cell proliferation.49 Thus, the inhibition of HER2 mutation leads to antitumor activity via decreased downstream cell-proliferation signaling.49 In 1998, trastuzumab was the first anti-HER2 monoclonal antibody to be approved by the FDA for the treatment of solid tumors.50 Trastuzumab is routinely used for the treatment of HER2-expressing cancers and is most frequently seen in breast cancer, gastric cancer, and esophageal or esophagogastric junction cancers, based on its benefits in overall and/or progression-free survival.28,51,52
In addition to trastuzumab, many anti-HER2 agents have been approved by the FDA, but only in the breast cancer setting. These agents include pertuzumab, ado-trastuzumab emtansine, fam-trastuzumab deruxtecan, lapatinib, neratinib, and tucatinib. The various anti-HER2 agents may differ in specific binding location (eg, intracellular, extracellular, and particular epitopes).
Cardiomyopathy, such as decreased left-ventricular ejection fraction, infusion reactions, and pulmonary adverse events are important monitoring points for most anti-HER2 therapies, especially intravenous agents. Although associated with intravenous agents, diarrhea and hepatotoxicity may occur more often with oral anti-HER2 agents, such as neratinib and lapatinib.53 In addition, ado-trastuzumab emtansine and fam-trastuzumab deruxtecan are antibody–drug conjugates that bind HER2, but they ultimately release a cytotoxic component (ie, DM1 and DXd, respectively), leading to bone marrow suppression.54,55
KIT Oncogene
The KIT proto-oncogene encodes the transmembrane RTK.56 KIT is activated through the binding of its ligand, stem-cell factor, which initiates downstream signaling via the Janus kinase (JAK)/signal transducer and activator of transcription (STAT), PI3K/Akt, and MAPK/ERK pathways.56,57 KIT mutations lead to the constitutive activation of downstream pathways, resulting in spontaneous proliferation and uncontrolled growth of tumor cells.56,57 KIT mutations are common driver mutations in gastrointestinal stromal tumors (GIST; 80%) and in melanoma (10%-15%).58,59
Mutations in KIT mainly affect the exons that encode the functional domains of the RTK, including exon 9, exon 11, exon 13, and exon 17.57 KIT exon 11 and exon 13 mutations have a high level of sensitivity to KIT inhibition, whereas KIT exon 9 and exon 17 mutations and KIT amplification have minimal or no sensitivity to KIT inhibitors.56,58
Imatinib, a BCR-ABL inhibitor, targets KIT and is FDA approved for the treatment of KIT (CD117)-positive GIST in the adjuvant or metastatic setting. The common adverse events associated with imatinib when used for the treatment of GIST include diarrhea, nausea, edema, and fatigue.60
KRAS Mutations
In contrast to NSCLC, EGFR testing is not recommended in patients with CRC, because it currently has no proved predictive value in determining a response to anti-EGFR monoclonal antibodies.61 Instead, the Kirsten rat sarcoma (KRAS) mutational status is used to identify appropriate patients to receive anti-EGFR monoclonal antibodies.61 KRAS mutations are strong, independent negative predictors of response to anti-EGFR monoclonal antibodies therapy.62 Approximately 40% of CRC cases are linked to KRAS mutations.63,64
In patients with KRAS wild-type advanced CRC, anti-EGFR monoclonal antibody therapy, such as cetuximab or panitumumab, may be added to standard chemotherapy to improve efficacy.65 These agents bind to EGFR, preventing phosphorylation and the activation of downstream kinases, which leads to tumor apoptosis.62
The common adverse events of cetuximab and panitumumab include hypersensitivity or infusion reactions, rashes or skin toxicities, and GI side effects.66
MET Inhibitors
The MET proto-oncogene encodes for the MET protein, a transmembrane RTK. MET RTK is activated via the binding of hepatocyte growth factor, leading to the downstream induction of the PI3K/Akt and MAPK/ERK pathways.67 Mutations and amplifications of MET and the overexpression of MET lead to the constitutive and prolonged activation of MET.68
The incidence of MET mutations is 5.6% in patients with NSCLC, 9% in patients with breast cancer, and up to 100% in papillary thyroid cancers.68,69 MET inhibition prevents the phosphorylation of RTK, thereby inhibiting the activation of those downstream pathways.70
Currently, the only FDA-approved MET-selective inhibitor is capmatinib, which is indicated for the treatment of metastatic NSCLC with MET exon 14 skipping mutation. Treatment with capmatinib may lead to peripheral edema and nausea most often; rare but serious side effects, including pneumonitis and hepatotoxicity, have been linked to treatment with capmatinib.71
Neurotrophic Tyrosine Receptor Kinase
The tropomyosin receptor kinase (Trk) family of receptors TrkA, TrkB, and TrkC are encoded by the neurotrophic tyrosine receptor kinase (NTRK)1, NTRK2, and NTRK3 genes. Trk proteins are transmembrane receptors, and through activation via ligand binding, signal transduction pathways leading to the modulation, proliferation, differentiation, and apoptosis of normal and neoplastic neuronal cells.72 The pathways involved include MAPK/ERK, PI3K/Akt, and phosphoinositide phospholipase C/protein kinase C (PLC/PKC).73,74
NTRK gene fusions are the most common genetic alteration with known oncogenic potential that lead to constitutively activated or overexpressed Trk.73,74 Chromosomal rearrangements involving the NTRK1, NTRK2, or NTRK3 genes occur in approximately 1% of all solid-tumor cancers.75 Entrectinib and larotrectinib inhibit Trk and are indicated for the treatment of solid tumors that express an NTRK gene fusion.75
Entrectinib and larotrectinib have favorable safety profiles. The common side effects of these drugs include fatigue and GI adverse events.76 In addition, on-target adverse events, such as dizziness, weight gain, and cognitive changes, can occur secondary to Trk inhibition in normal tissues.76
PIK3CA Mutations
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutations are the oncogenic driver in a variety of cancers.77 Alterations in this pathway most frequently result from somatic mutations in the PIK3CA coding p110α or PIK3CA amplification and can lead to enhanced cell growth, antiapoptosis, cell-cycle progression, and translation.77,78
The PIK3CA-activating mutations occur in approximately 40% of HR-positive breast cancer.79 Alpelisib, a PI3K inhibitor, is approved, in combination with fulvestrant, after disease progression during or after receiving an endocrine-based regimen, for the treatment of postmenopausal women with HR-positive advanced or metastatic breast cancer and PIK3CA mutation. This approval was based on data demonstrating a progression-free survival benefit with alpelisib compared with placebo.79 The adverse events associated with alpelisib include rash, GI adverse events, hyperglycemia, hypersensitivity reactions, and pulmonary side effects.79
PDGFRA Mutations
Platelet-derived growth factor receptor alpha (PDGFRA) mutations are oncogenic drivers in 5% to 10% of GIST cases.80 The specific PDGFRA mutation D842V occurs in 5% to 6% of GIST cases and is resistant to currently available tyrosine kinase inhibitors that target PDGFRA other than avapritinib.81 Avapritinib was approved in January 2020 for the treatment of patients with GIST and a PDGFRA exon 18 mutation, including PDGFRA D842V mutations.81
Avapritinib inhibits PDGFRA, which prevents the autophosphorylation of PDGFRA, thus preventing tumor proliferation. The adverse events of avapritinib may include cognitive changes, nausea, vomiting, diarrhea, and edema.81
RET Oncogene
The rearranged during transfection (RET) is a proto-oncogene that encodes a transmembrane protein that is involved in cell growth via multiple pathways, including MAPK/ERK, PI3K/Akt, JAK/STAT3, and PLC/PKC. RET mutations and fusions are oncogenic drivers in NSCLC and in thyroid cancer.82
Approximately 1% to 2% of patients with NSCLC have RET fusions, with KIF5B-RET being the most common.82 RET fusions occur in 20% to 40% of papillary thyroid cancers.83 In addition, RET mutations occur in 43% to 71% of patients with sporadic medullary thyroid cancer, whereas germline RET mutations occur in 88% of familial medullary thyroid cancers and 95% of multiple neoplasia type 2A cases.83,84
The first 2 FDA-approved RET inhibitors were selpercatinib and pralsetinib, which are indicated for the treatment of patients with advanced or metastatic medullary thyroid cancer and RET mutation, advanced or metastatic thyroid cancer with RET fusion, and metastatic NSCLC with RET fusion.
The drug-class adverse events include hepatotoxicity, hypertension, hemorrhage, and GI adverse events.85,86
ROS1 Oncogene
The rearrangement of the ROS1 gene leads to a constitutively activated RTK, which results in dysregulation and inappropriate cell signaling via the MAPK/ERK pathway.87 ROS1 rearrangements were initially identified in glioblastoma cells; in 2007 ROS1 rearrangements were first discovered in NSCLC.88,89
ROS1 rearrangements occur in 1% to 2% of patients with NSCLC, and the incidence is higher in nonsmokers and in patients with adenocarcinoma histology.87,90,91
Because of the structural similarities between ROS1 and ALK within the kinase domain and the ATP-binding sites, many agents that inhibit ALK rearrangements also inhibit the ROS1 mutations.92,93 ALK inhibitors (crizotinib) and NTRK inhibitors (entrectinib) have been approved by the FDA for patients with metastatic NSCLC and ROS1 mutation and have improved the overall response rate, progression-free survival, and/or overall survival.93-95
Conclusion
As the number of actionable biomarkers continues to increase, testing patients with solid tumors for targetable biomarkers is critically important, to guide the selection of available treatment options. Predictive biomarker-driven targeted therapies will continue to expand to provide new treatment options for patients, which will subsequently improve patient outcomes and treatment safety. A potential targeted therapy class that may soon be approved by the FDA is KRAS G12C inhibitors, which shows promising efficacy in patients with KRAS G12C mutations. In addition, the indications of the currently approved treatments may be expanded in the future. Within this rapidly growing clinical area, oncology pharmacists are essential in managing the treatment selection, drug interactions, and treatment side effects.
Author Disclosure Statement
Dr Mo and Dr Renna have no conflicts of interest to report.
References
- Hockings JK, Pasternak AL, Erwin AL, et al. Pharmacogenomics: an evolving clinical tool for precision medicine. Cleve Clin J Med. 2020;87:91-99.
- Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526:343-350.
- Khan M, Lin J, Liao G, et al. ALK inhibitors in the treatment of ALK positive NSCLC. Front Oncol. 2019;8:557. doi.org/10.3389/fonc.2018.00557.
- Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4:1450-1454.
- Rothenstein JM, Letarte N. Managing treatment–related adverse events associated with Alk inhibitors. Curr Oncol. 2014;21:19-26.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Non-Small Cell Lung Cancer. Version 2.2021. December 15, 2020. www.nccn.org/professionals/physician_gls/pdf/nscl.pdf. Accessed January 27, 2021.
- Nakagawa K, Hida T, Nokihara H, et al. Final progression-free survival results from the J-ALEX study of alectinib versus crizotinib in ALK-positive non-small-cell lung cancer. Lung Cancer. 2020;139:195-199.
- Solomon BJ, Besse B, Bauer TM, et al. Lorlatinib in patients withALK-positive non-small-cell lung cancer: results from a global phase 2 study. Lancet Oncol. 2018;19:1654-1667.
- Sanchez JN, Wang T, Cohen MS. BRAF and MEK inhibitors: use and resistance in BRAF-mutated cancers. Drugs. 2018;78:549-566.
- Li Y, Umbach DM, Li L. Putative genomic characteristics of BRAF V600K versus V600E cutaneous melanoma. Melanoma Res. 2017;27:527-535.
- Hugdahl E, Kalvenes MB, Puntervoll HE, et al. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br J Cancer. 2016;114:801-808.
- Gogas H, Ascierto PA, Flaherty K, et al. Update on overall survival in COLUMBUS: A randomized phase III trial of encorafenib (ENCO) plus binimetinib (BINI) versus vemurafenib (VEM) or ENCO in patients with BRAF V600-mutant melanoma. J Clin Oncol. 2020;38(15 suppl):Abstract 10012.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Korphaisarn K, Kopetz S. BRAF-directed therapy in metastatic colorectal cancer. Cancer J. 2016;22:175-178.
- Heinzerling L, Eigentler TK, Fluck M, et al. Tolerability of BRAF/MEK inhibitor combinations: adverse event evaluation and management. ESMO Open. 2019;4:e000491. doi.org/10.1136/esmoopen-2019-000491.
- Ducreux M, Chamseddine A, Laurent-Puig P, et al. Molecular targeted therapy of BRAF-mutant colorectal cancer. Ther Adv Med Oncol. 2019;11:1758835919856494. doi.org/10.1177/1758835919856494.
- Subbiah V, Kreitman RJ, Wainberg ZA, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600–mutant anaplastic thyroid cancer. J Clin Oncol. 2018;36:7-13.
- Bustamante Alvarez JG, Otterson GA. Agents to treat BRAF-mutant lung cancer. Drugs Context. 2019;8:212566. DOI: 10.7573/dic.212566. Accessed March 1, 2021.
- Pilié PG, Gay CM, Byers LA, et al. PARP inhibitors: extending benefit beyond BRCA-mutant cancers. Clin Cancer Res. 2019;25:3759-3771.
- Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1274-1284.
- Robson M, Im SA, Senkus E, Xu B, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523-533.
- Golan T, Hammel P, Reni M, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317-327.
- Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753-763.
- Ramus SJ, Gayther, SA. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol. 2009;3:138-150.
- Messina C, Cattrini C, Soldato D, et al. BRCA mutations in prostate cancer: prognostic and predictive implications. J Oncol. 2020;2020:4986365. doi.org/10.1155/2020/4986365.
- LaFargue CJ, Dal Molin GZ, Sood AK, Coleman RL. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019;20:e15-e28. doi.org/10.1016/S1470-2045(18)30786-1.
- National Cancer Institute. Cancer stat facts: female breast cancer subtypes. https://seer.cancer.gov/statfacts/html/breast-subtypes.html. Accessed February 1, 2020.
- Gradishar WJ, Anderson BO, Abraham J, et al. Breast cancer, version 3.2020. NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2020;18:452-478.
- Francis PA, Pagani O, Fleming GF, et al.Tailoring adjuvant endocrine therapy for premenopausal breast cancer. N Engl J Med. 2018;379:122-137.
- Miller WR. Aromatase inhibitors: mechanism of action and role in the treatment of breast cancer. Semin Oncol. 2003;30(4 suppl 14):3-11.
- Scott SC, Lee SS, Abraham J. Mechanisms of therapeutic CDK4/6 inhibition in breast cancer. Semin Oncol. 2017;44:385-394.
- Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77.
- Turner NC, Slamon DJ, Ro J, et al. Overall survival with palbociclib and fulvestrant in advanced breast cancer. N Engl J Med. 2018;379:1926-1936.
- Sledge GW Jr, Toi M, Neven P, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2– advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35:2875-2884.
- Im SA, Lu YS, Bardia A, et al. Overall survival with ribociclib plus endocrine therapy in breast cancer. N Engl J Med. 2019;381:307-316.
- Cazzaniga ME, Danesi R, Girmenia C, et al. Management of toxicities associated with targeted therapies for HR-positive metastatic breast cancer: a multidisciplinary approach is the key to success. Breast Cancer Res Treat. 2019;176:483-494.
- Hirsch FR, Bunn PA Jr. EGFR testing in lung cancer is ready for prime time. Lancet Oncol. 2009;10:432-433.
- Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378:113-125.
- Ahn MJ, Tsai CM, Shepherd FA, et al. Osimertinib in patients with T790M mutation-positive, advanced non-small cell lung cancer: long-term follow-up from a pooled analysis of 2 phase 2 studies. Cancer. 2019;125:892-901.
- Takeda M, Nakagawa K. Toxicity profile of epidermal growth factor receptor tyrosine kinase inhibitors in patients with epidermal growth factor receptor gene mutation-positive lung cancer. Mol Clin Oncol. 2017;6:3-6.
- Haugsten EM, Wiedlocha A, Olsnes S, Wesche J. Roles of fibroblast growth factor receptors in carcinogenesis. Mol Cancer Res. 2010;8:1439-1452.
- Ross JS, Wang K, Khaira D, et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer. 2016;122:702-711.
- Loriot Y, Necchi A, Park SH, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. N Engl J Med. 2019;381:338-348.
- Krook MA, Lenyo A, Wilberding M, et al. Efficacy of FGFR inhibitors and combination therapies for acquired resistance in FGFR2-fusion cholangiocarcinoma. Mol Cancer Ther. 2020;19:847-857.
- Chae YK, Ranganath K, Hammerman PS, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. 2017;8:16052-16074.
- Ray-Coquard I, Pautier P, Pignata S, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381:2416-2428.
- Moore KN, Secord AA, Geller MA, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20:636-648.
- de Bono J, Mateo J, Fizazi K, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382:2091-2102.
- Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11:263-275.
- Dillman RO. Perceptions of herceptin: a monoclonal antibody for the treatment of breast cancer. Cancer Biother Radiopharm. 1999;14:5-10.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): gastric cancer. Version 4.2020. December 23, 2020. www.nccn.org/professionals/physician_gls/pdf/gastric.pdf. Accessed February 1, 2021.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): esophageal and esophagogastric junction cancers. Version 5.2020. December 23, 2020. www.nccn.org/professionals/physician_gls/pdf/esophageal.pdf. Accessed February 1, 2021.
- Xuhong JC, Qi XW, Zhang Y, Jiang J. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am J Cancer Res. 2019;9:2103-2119.
- Burris 3rd HA, Rugo HS, Vukelja SJ, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398-405.
- Modi S, Saura C, Yamashita T, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382:610-621.
- Liang J, Wu YL, Chen BJ, et al. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int J Biol Sci. 2013;9:435-443.
- Martín-Broto J, Rubio L, Alemany R, Lopez-Guerrero JA. Clinical implications of KIT and PDGFRA genotyping in GIST. Clin Transl Oncol. 2010;12:670-676.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): cutaneous melanoma. Version 1.2021. November 25, 2020. www.nccn.org/professionals/physician_gls/pdf/cutaneous_melanoma.pdf. Accessed February 1, 2021.
- Joensuu H. Gastrointestinal stromal tumor (GIST). Ann Oncol. 2006;17 Suppl 10:x280-x286.
- Schlemmer M, Bauer S, Schütte R, et al. Activity and side effects of imatinib in patients with gastrointestinal stromal tumors: data from a German multicenter trial. Eur J Med Res. 2011;16:206-212.
- Hecht JR, Mitchell E, Neubauer MA, et al. Lack of correlation between epidermal growth factor receptor status and response to panitumumab monotherapy in metastatic colorectal cancer. Clin Cancer Res. 2010;16:2205-2213.
- Lièvre A, Bachet JB, Le Corre D, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992-3995.
- Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol. 2010;28:466-474.
- Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626-1634.
- Van Cutsem E, Kohne CH, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009;360:1408-1417.
- Petrelli F, Ardito R, Ghidini A, et al. Different toxicity of cetuximab and panitumumab in metastatic colorectal cancer treatment: a systematic review and meta-analysis. Oncology. 2018;94:191-199.
- Organ SL, Tsao MS. An overview of the C-MET signaling pathway. Ther Adv Med Oncol. 2011;3:S7-S19.
- Tovar EA, Graveel CR. MET in human cancer: germline and somatic mutations. Ann Transl Med. 2017;5:205.
- Trovato M, Campenni A, Giovinazzo S, et al. Hepatocyte growth factor/C-Met axis in thyroid cancer: from diagnostic biomarker to therapeutic target. Biomark Insights. 2017;12:1177271917701126.
- Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017;3:148-153.
- Wolf J, Seto T, Han JY, et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N Engl J Med. 2020;383:944-957.
- Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169:107-114.
- Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open. 2016;1:e000023.
- Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25-34.
- Ricciuti B, Genova C, Crino L, Libra M, Leonardi GC. Antitumor activity of larotrectinib in tumors harboring NTRK gene fusions: a short review on the current evidence. OncoTargets Ther. 2019;12:3171-3179.
- Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30(suppl_8):viii23-viii30.
- Mukohara T. PI3K mutations in breast cancer: prognostic and therapeutic implications. Breast Cancer (Dove Med Press). 2015;7:111-123.
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase (PI3K) pathway in cancer. Nat Rev Drug Discov. 2009;8:627-644.
- André F, Ciruelos E, Rubovszky G, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. 2019;380:1929-1940.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): gastrointestinal stromal tumors (GISTs). Version 1.2021. October 10, 2020. www.nccn.org/professionals/physician_gls/pdf/gist.pdf. Accessed February 25, 2021.
- Henrich M, Jones R, Mehren M, et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): a multicentre, open-label, phase 1 trial. Lancet Oncol. 2020;21:935-946.
- Drilon A, Wang L, Hasanovic A, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 2013;3:630-635.
- Kato S, Subbiah V, Marchlik E, et al. RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin Cancer Res. 2017;23:1988-1997.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): thyroid carcinoma. Version 2.2020. July 15, 2020. www.nccn.org/professionals/physician_gls/pdf/thyroid.pdf. Accessed February 1, 2021.
- Subbiah V, Hu MI, Gainor JF, et al. Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J Clin Oncol. 2020;38(15 suppl):Abstract 109.
- Wirth LJ, Sherman E, Robinson B, et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N Engl J Med. 2020;383:825-835.
- Rossi G, Jocolle G, Conti A, et al. Detection of ROS1 rearrangement in non-small cell lung cancer: current and future perspectives. Lung Cancer (Auckl). 2017;8:45-55.
- Birchmeier C, Sharma S, Wigler M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc Natl Acad Sci USA. 1987;84:9270-9274.
- Gainor JF, Shaw AT. Novel targets in non-small cell lung cancer: ROS1 and RET fusions. Oncologist. 2013;18:865-875.
- Dugay F, Llamas-Gutierrez F, Gournay M, et al. Clinicopathological characteristics of ROS1- and RET-rearranged NSCLC in caucasian patients: data from a cohort of 713 non-squamous NSCLC lacking KRAS/EGFR/HER2/BRAF/PIK3CA/ALK alterations. Oncotarget. 2017;8:53336-53351.
- Bergethon K, Shaw AT, Ou SH, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863-870.
- Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non–small-cell lung cancer. N Engl J Med. 2014;371:1963-1971.
- Chin LP, Soo RA, Soong R, Ou SH. Targeting ROS1 with anaplastic lymphoma kinase inhibitors: a promising therapeutic strategy for a newly defined molecular subset of non–small-cell lung cancer. J Thorac Oncol. 2012;7:1625-1630.
- Drilon A, Siena S, Dziadziuszko R, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21:261-270.
- Lim SM, Kim HR, Lee JS, et al. Open-label, multicenter, phase II study of ceritinib in patients with non–small-cell lung cancer harboring ROS1 rearrangement. J Clin Oncol. 2017;35:2613-2618.