Non-Hodgkin lymphoma (NHL) includes a heterogenous assortment of lymphoproliferative malignancies arising from aberrant B lymphocytes, T lymphocytes, or natural killer cells. According to the World Health Organization, NHL has more than 60 histopathologic subtypes, with approximately 90% originating from B lymphocytes.1
The most common B-cell lymphoma subtypes include Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), mantle-cell lymphoma (MCL), and marginal-zone lymphoma.
The overall prognosis and treatment for B-cell lymphoma is multifactorial and highly variable, and are contingent on disease classification, staging, genetic features, and other patient-specific factors.2 Immunotherapy, which is now considered to be a pillar of cancer treatment, has significantly improved overall survival (OS) in various NHL subtypes.3
Immune effector cell therapy, also known as adoptive cell therapy, is an innovative approach to cancer treatment that uses genetically engineered autologous T-cells. In 2017, tisagenlecleucel (Kymriah) became the first-in-class anti-CD19 chimeric antigen receptor (CAR) T-cell therapy to be approved by the US Food and Drug Administration (FDA), 30 years after the initial conception of a CAR.4,5 After this groundbreaking approval, gene therapy has continued to evolve and gain significant traction in the treatment of common B-cell lymphomas, acute lymphoblastic leukemia (ALL), and other hematologic malignancies.6
Currently, 4 CAR T-cell agents are FDA-approved specifically for the treatment of various relapsed or refractory B-cell lymphomas.4,7-9 Tisagenlecleucel and lisocabtagene maraleucel (Breyanzi) are indicated for the treatment of relapsed or refractory DLBCL.4,9 Axicabtagene ciloleucel (Yescarta) is also indicated for the treatment of relapsed or refractory DLBCL, and is the first and currently only CAR T-cell agent approved for the treatment of relapsed or refractory FL.7 The fourth agent, brexucabtagene autoleucel (Tecartus) is the sole CAR T-cell therapy approved for the treatment of relapsed or refractory MCL.8
The aim of this review was to evaluate the safety, efficacy, and practical considerations for these 4 revolutionary agents in the management of patients with relapsed or refractory B-cell lymphomas.
Composition of CAR T-Cell Therapies
In 1909, the Nobel Laureate Paul Ehrlich famously hypothesized that immunosurveillance mechanisms might prevent tumorigenesis.10 This now widely recognized concept became the foundation for immunotherapy and, eventually, for the creation of adoptive gene therapy. CAR T-cell therapy is a unique immunologic approach to cancer treatment that effectively transforms endogenous T-cells into “living drugs” that may persist in their anticancer activity for months or years after administration.11
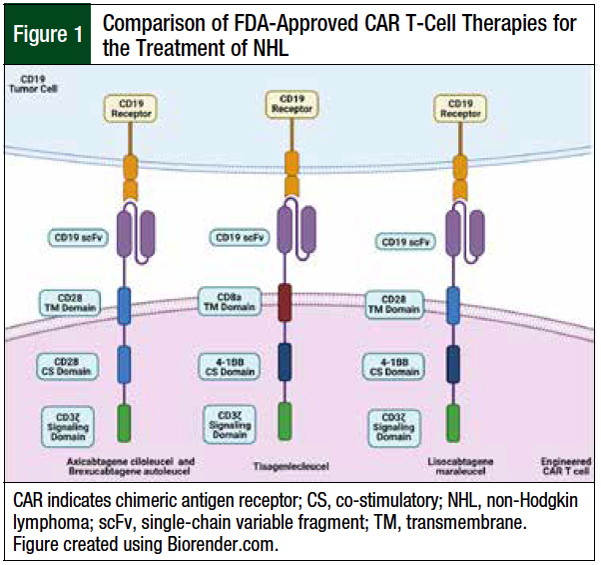
Autologously obtained T-cells are genetically altered ex vivo via lentiviral transduction to express a murine single-chain variable fragment CAR protein construct with redirected antitumor specificity.6 All current CAR T-cell drugs approved for the treatment of B-cell lymphomas contain anti-CD19 antigen–binding domains.4,7-9 To date, no other antigen has shown results equal or superior to CD19 in the treatment of B-cell lymphomas.12
The first-generation CAR T-cell therapies lacked co-stimulatory domains and had poor efficacy and persistence in early phase 1 clinical trials, leading researchers to create the second-generation CAR T-cell therapies, which are containing an extracellular antigen recognition domain, a transmembrane domain, and a co-stimulatory domain attached to the cytoplasmic tail of the essential CD3ζ intracellular-signaling domain.12,13
Figure 1 highlights the various compositions of the current 4 FDA-approved CAR T-cell therapies for the treatment of B-cell lymphomas.
Pharmacology of CAR T-Cell Therapies
Designed to seek out and engage CD19-expressing B lymphocytes, the current anti-CD19 CAR T-cell therapies indiscriminately attack healthy and malignant B lymphocytes.14 Once bound to the CD19 cell surface epitope, the CAR T-cell becomes activated in a major histocompatibility complex–independent manner.14
After T-cell activation, intracellular signal transduction from the co-stimulatory domain, notably CD28 or 4-1BB, is responsible for enhanced cytotoxicity, T-cell proliferation, cytokine release, and a sustained antitumor response.12,15
The second-generation anti-CD19 CAR T-cell therapies use various co-stimulatory and transmembrane domains; however, the optimal pairing and construct of these 2 domains remain unknown and require additional investigation.
Manufacturing of CAR T-Cells
As an autologous cell–based therapy, CAR T-cell manufacturing is a multistep process that involves (1) harvesting peripheral blood mononuclear cells via leukapheresis, (2) shipping the cryopreserved apheresis drug to a manufacturing site, (3) genetically modifying and expanding T-cells ex vivo, (4) having process control checkpoints to ensure sufficient CAR T-cell quality and quantity, and (5) shipping the final drug back to the treatment center for infusion (Figure 2).16
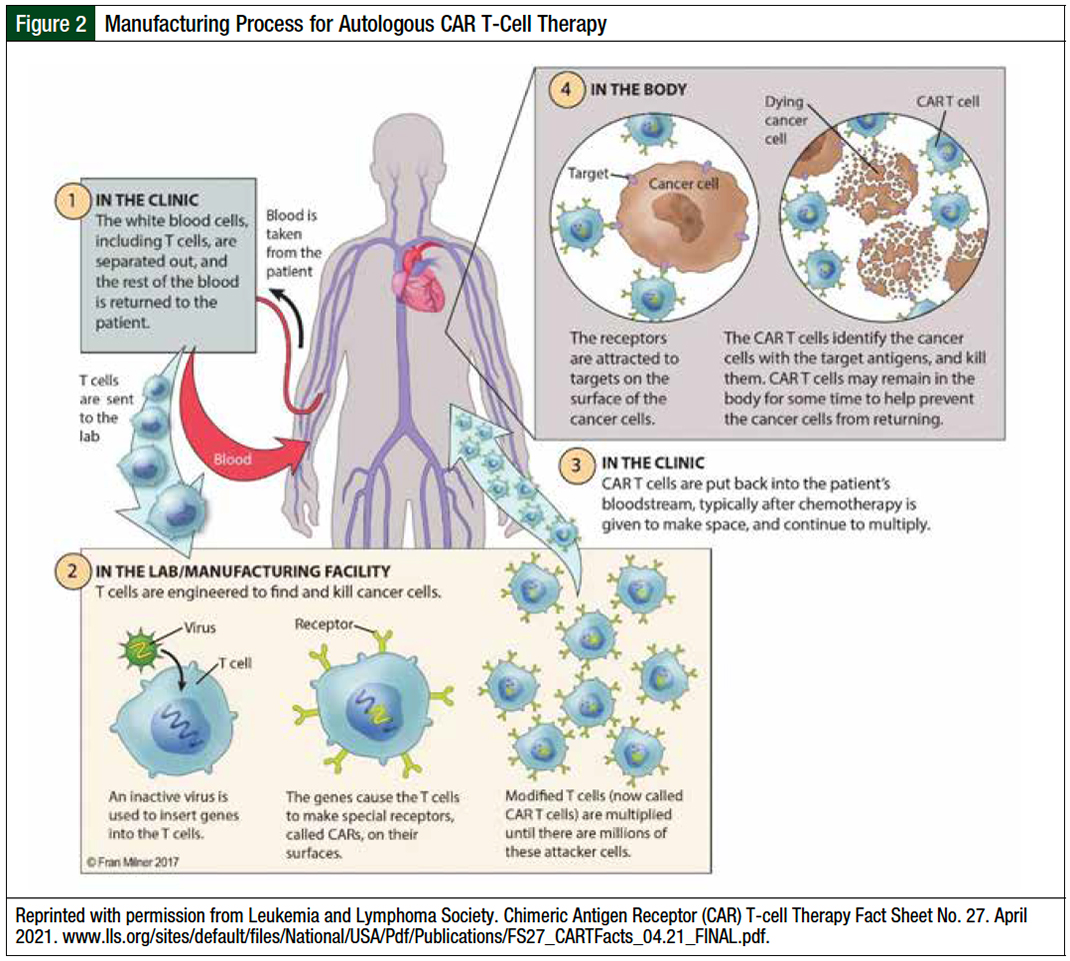
The manufacturing times for CAR T-cells varied in pivotal phase 2 clinical trials. The median time from leukapheresis to the infusion of axicabtagene ciloleucel and brexucabtagene autoleucel were 24 days (range, 16-73 days) and 27 days (range, 19-63 days), respectively.7,8 The median time from leukapheresis to the infusion of lisocabtagene maraleucel was 37 days (range, 27-224 days).17 Finally, the median time from leukapheresis to the infusion of tisagenlecleucel in patients with DLBCL was 113 days (range, 47-196 days).4
Axicabtagene ciloleucel and brexucabtagene autoleucel are similar constructs produced by the same manufacturer; however, brexucabtagene autoleucel involves a unique and patented XLP process that facilitates the removal of circulating CD19-expressing tumor cells after apheresis.18 This additional step may minimize the likelihood of premature activation, expansion, and exhaustion of anti-CD19 CAR T-cells during ex vivo manufacturing.
Dosing and Administration of CAR T-Cells
Lymphodepleting chemotherapy administered before CAR T-cell infusion reduces endogenous lymphocytes and increases anti-CD19 CAR T-cell response.19 The ideal conditioning regimen is unknown; however, many institutions and clinical trials prefer fludarabine and cyclophosphamide treatment for 3 days before the infusion of CAR T-cells.
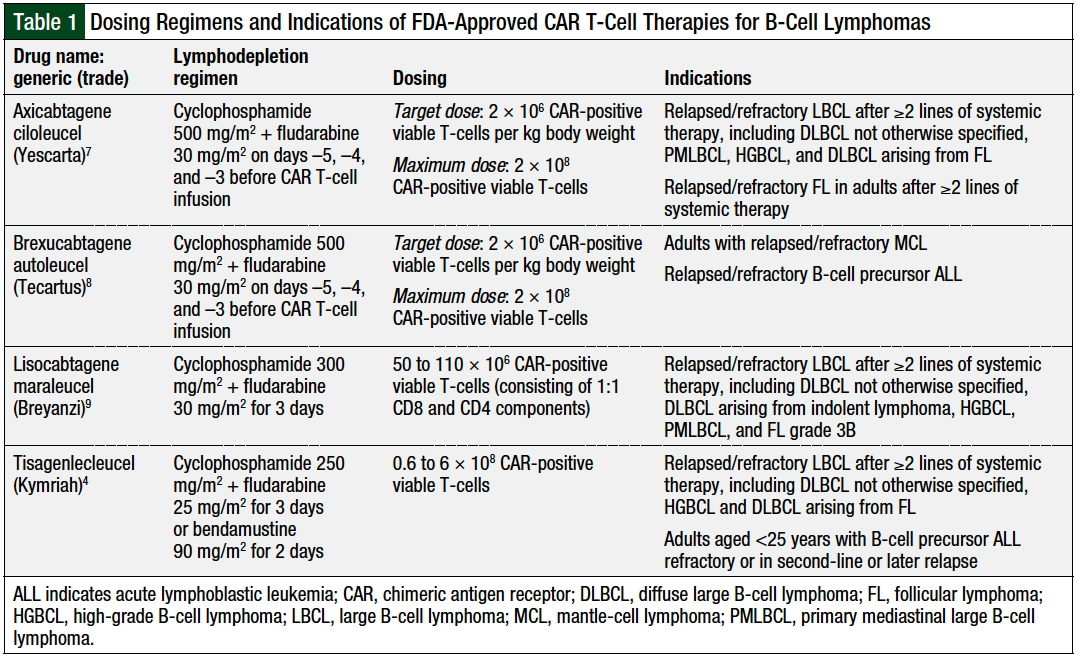
Bridging modalities—including systemic chemotherapy, corticosteroids, oral oncolytics, radiation, or immunotherapy—may be warranted to help control disease while the patient awaits the manufacturing and delivery of CAR T-cell therapies.20 Each CAR T-cell therapy has a different target CAR-positive viable T-cell dose, with actual cell counts and volumes varying in all final patient-specific drugs (Table 1).
Clinical Efficacy in B-Cell Lymphomas
Following the pioneering work of Eshhar and colleagues,21 Rosenberg and colleagues,22 and others,23,24 sentinel early-phase clinical trials with anti-CD19 CAR T-cells were initiated in patients with ALL.25,26 In 2010, researchers at the National Cancer Institute treated the first patient with B-cell lymphoma with genetically modified autologous T-cells.27 Such investigations resulted in several second-generation constructs that merited further evaluation in phase 2 clinical trials.
Axicabtagene Ciloleucel
In the pivotal, single-arm, multicenter, open-label, phase 1/2 ZUMA-1 clinical trial, axicabtagene ciloleucel was evaluated in 111 adults with refractory large B-cell lymphoma (LBCL).28 Axicabtagene ciloleucel was effectively manufactured for 110 (99%) of these patients and was administered to 101 (91%) patients. For the phase 2 component of ZUMA-1, the objective response rate (ORR), the study primary end point, was 82%, including 54% complete responses. After a median follow-up of 15.4 months, 40% of patients with a complete response had ongoing responses, and the estimated OS rates at 12 and 18 months were 59% and 52%, respectively.28
On October 18, 2017, axicabtagene ciloleucel was the first gene-based therapy to receive FDA approval for the treatment of patients with relapsed or refractory LBCL (after ≥2 lines of therapy).29
The updated analysis of ZUMA-1, with a median follow-up of 27.1 months (interquartile range, 25.7-28.8), included all 101 evaluable patients who received axicabtagene ciloleucel in phase 2 of the study.30 Similar to the original ZUMA-1 analysis, the ORR and the complete response rate were relatively stable at 83% and 58%, respectively. The overall median duration of response (DOR) was 11.1 months (range, 4.2 months-not estimable), but the median DOR for the patients with a complete response had not been reached.30
At data cutoff, 39 (39%) patients had ongoing responses, with 37 of them having a complete response. The median progression-free survival (PFS) was 5.9 months (95% confidence interval [CI], 3.3-15), and 61 of the 101 (60%) patients had disease progression or died during the study. The study showed that higher response rates were associated with increased CAR T-cell peak concentrations and with increased area under the curve in the first 28 days after the administration of axicabtagene ciloleucel.30
A 4-year analysis of 101 evaluable patients, with a median follow-up of 51.1 months, showed a median time to next therapy of 8.7 months (range, 0.3-53.8 months) and a median OS of 25.8 months.31 This long-term follow-up of ZUMA-1 showed deep and durable responses, with an estimated 4-year OS rate of 44%.31
ZUMA-5 was a single-arm, open-label, multicenter, phase 2 clinical trial that led to the FDA approval of axicabtagene ciloleucel for the treatment of relapsed or refractory nontransformed FL.32 The interim analysis included 84 patients with relapsed or refractory FL and at least 12 months of follow-up. Before enrollment, these patients had received a median of 3 previous lines of therapy (range, 1-10), including 64% who received ≥3 lines. The ORR in these heavily pretreated patients was 94%, including 80% complete responses. At the data cutoff at a median follow-up of 17.5 months, 64% of patients had an ongoing response. For all patients with indolent NHL who were evaluated, the 12-month estimated OS, PFS, and disease-free survival rates were 93% (95% CI, 86-97), 74% (95% CI, 63-82), and 72% (95% CI, 61-80), respectively.32 Axicabtagene ciloleucel is currently the first and only CAR T-cell therapy approved for the treatment of relapsed or refractory nontransformed FL after ≥2 lines of systemic therapy.33
Tisagenlecleucel
On August 30, 2017, the FDA approved tisagenlecleucel for the treatment of patients aged ≤25 years with relapsed or refractory ALL.34 With notable anti-CD19 activity, the investigators then evaluated tisagenlecleucel therapy in the JULIET study for the treatment of adults with relapsed or refractory DLBCL after ≥2 lines of therapy, including an anthracycline and rituximab.35 The pivotal JULIET study was an international, single-arm, open-label, phase 2 clinical trial of 115 patients between July 2015 and December 2017.35
The primary efficacy analysis included 93 patients who had received tisagenlecleucel, and had ≥3 months of follow-up or who discontinued the study before 3 months. The ORR was 52% (95% CI, 41-62), including 40% complete responses and 12% partial responses. For responders, the estimated 12-month relapse-free survival rate was 65%, and the OS rate was 90% for those with a complete response (49% among all patients).35
The DOR for patients with a complete response was not reached (95% CI, 10 months-not reached). Of note, 50 (30%) patients discontinued the study before receiving the CAR T-cell therapy, mainly because of disease progression; death; or manufacturing failure in 12 patients.35 Based on the high durable response rates in the JULIET study, on May 1, 2018, the FDA approved tisagenlecleucel for the treatment of adults with relapsed or refractory LBCL after ≥2 lines of therapy.36
Chong and colleagues evaluated the 5-year outcomes after tisagenlecleucel administration in 38 patients with DLBCL (N = 24) or with FL (N = 14).37 The study’s median follow-up was 60.7 months. For the 24 patients with DLBCL, the ORR was 58%, including 11 (46%) patients with complete responses. At 5 years, the PFS was 31% (95% CI, 14-51) and the median DOR was 61.4 months (95% CI, 3.2-not estimable). For the 14 patients with FL, the ORR was 79%, including 71% complete responses.37
The 5-year PFS was 43% (95% CI, 18-66), and the median DOR had not been reached (95% CI, 9.5 months-not estimable).37 An important aspect of this study was the assessment of CAR T-cell persistence. The CAR19 transgene was detectable by polymerase chain reaction assay throughout the follow-up period in 6 (50%) of the 12 patients with a complete response beyond 1 year (median duration, 39.4 months; range, 22.5-57.3).37 This 5-year analysis is the longest follow-up of any anti-CD19 CAR T-cell agent to date.
Brexucabtagene Autoleucel
The pivotal, single-arm, international, open-label, phase 2 ZUMA-2 clinical trial included 74 adults with relapsed or refractory MCL who received brexucabtagene autoleucel.38 Patients must have not responded to therapy with at least an anthracycline- or bendamustine-containing chemotherapy regimen, a Bruton tyrosine kinase (BTK) inhibitor, and an anti-CD20 monoclonal antibody. The patients also had to have either cyclin D1 overexpression or translocation t(11;14), which are key pathogenic events in MCL and result in cell-cycle dysregulation and hyperproliferation.39 Of the 74 patients who underwent leukapheresis, 68 received brexucabtagene autoleucel after lymphodepleting chemotherapy, and 6 patients did not receive the CAR T-cell therapy, because of manufacturing failure (N = 3); progressive disease (N = 2); or complications from lymphodepleting chemotherapy (N = 1).38
A primary efficacy analysis was reported for the first 60 patients who had a 7-month evaluation after CAR T-cell therapy administration.38 The Independent Review Committee reported an ORR of 93% (95% CI, 84-98), including 67% (95% CI, 53-78) complete responses. An intention-to-treat analysis for all 74 patients showed an ORR of 85%, including 59% complete responses. With a median follow-up of 12.3 months (range, 7-32.3 months), 57% of patients in the primary efficacy cohort remained in remission. The estimated 12-month OS and PFS rates were 83% and 61%, respectively.38 On July 24, 2020, the FDA granted an accelerated approval to brexucabtagene autoleucel as the first-in-class CAR T-cell therapy for the treatment of adults with relapsed or refractory MCL.40
Wang and colleagues conducted an updated analysis of the safety and efficacy of brexucabtagene autoleucel in patients with relapsed or refractory MCL.41 With a median follow-up of 17.5 months (range, 12.3-37.6 months), the ORR and complete response rate in 60 efficacy-evaluable patients were 92% (95% CI, 81.6-97.2) and 67% (95% CI, 53.3-78.3), respectively. The median OS, median PFS, and median DOR were not reached; however, the 15-month estimated OS, PFS, and DOR rates were 76% (95% CI, 62.8-85.1), 59.2% (95% CI, 44.6-71.2), and 58.6% (95% CI, 42.5-71.7), respectively. Overall, 48% of the efficacy-evaluable patients had ongoing clinical responses at the data cutoff of December 31, 2019.41
Lisocabtagene Maraleucel
The multicenter, multicohort, open-label, phase 2 TRANSCEND NHL 001 clinical trial enrolled 344 adults with relapsed or refractory LBCL.17 Of the patients who underwent leukapheresis, 269 (78%) received at least 1 dose of lisocabtagene autoleucel, 25 received a nonconforming (ie, safe for infusion but not meeting the manufacturer’s criteria for drug administration) CAR T-cell therapy, 2 patients did not receive CAR T-cells because of manufacturing failure, and 48 patients were ineligible for the study because of lymphoma complications and/or death before CAR T-cell administration. Among the efficacy-evaluable 256 patients, the ORR was 73% (95% CI, 66.8-78), including 53% (95% CI, 46.8-59.4) complete responses.
The median follow-up for all 344 patients who underwent leukapheresis was 18.8 months (95% CI, 15-19.3 months). The estimated 12-month OS was 58% for the entire population, with a median OS of 21.1 months (95% CI, 13.3 months-not reached) in the efficacy-evaluable cohort. With a median follow-up of approximately 12 months, the median DOR was not reached (95% CI, 8.6 months-not reached), and the median PFS was 6.8 months (95% CI, 3.3-14.1 months).17 Based on these results, on February 5, 2021, the FDA granted accelerated approval to lisocabtagene maraleucel for the treatment of adults with relapsed or refractory LBCL who received ≥2 previous lines of therapy.42
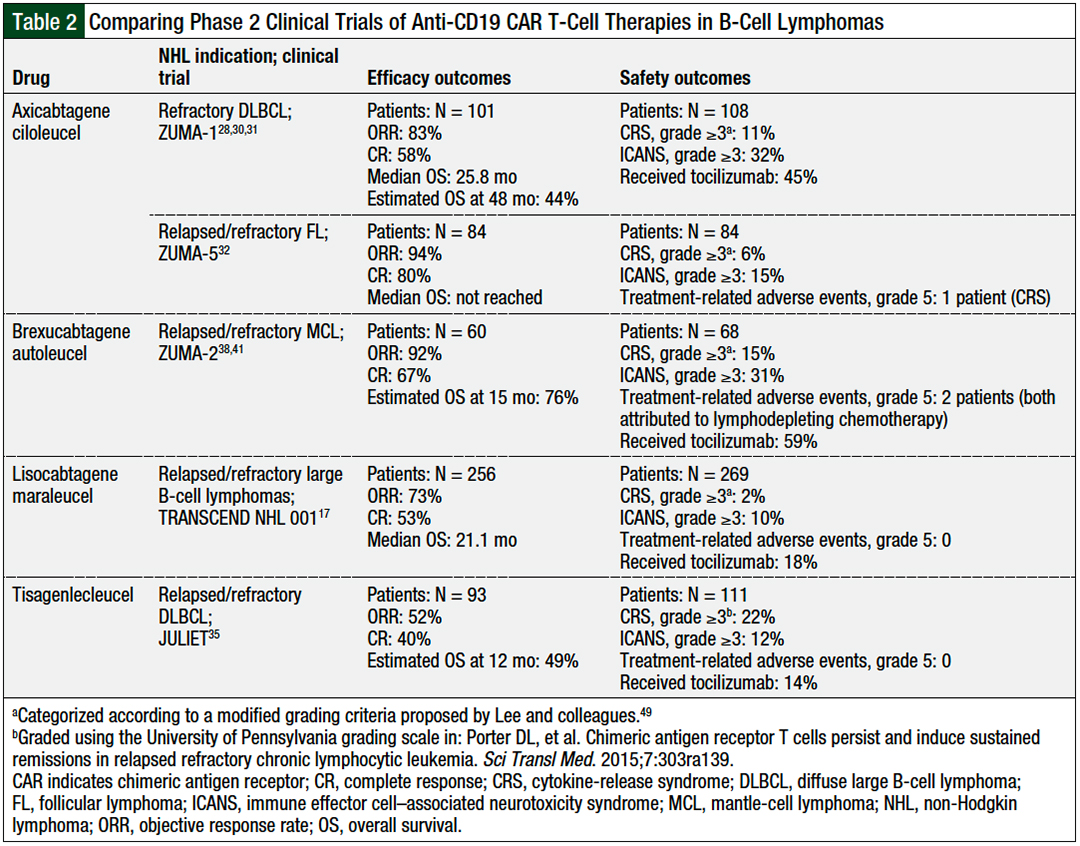
Table 2 provides a comparison of the efficacy and safety outcomes of phase 2 clinical trials of the CAR T-cell therapies used in B-cell lymphomas.
Comparison of CAR T-Cell Therapies
SCHOLAR-1 was an international, retrospective analysis of patients with relapsed or refractory DLBCL from 2 large randomized clinical trials and 2 academic databases.43 A total of 636 patients were included in the analysis, which aimed to illuminate the response rates and median OS in patients with refractory DLBCL. Using conventional salvage options, the ORR was 26% and the median OS was 6.3 months, highlighting the unmet medical need in this high-risk patient population. SCHOLAR-1 shows the exigency for innovative treatment options43; however, this study is not an ideal analysis to evaluate alongside the previously mentioned pivotal phase 2 anti-CD19 CAR T-cell clinical trials.17,28,35,38
Furthermore, cross-trial comparisons of small, single-arm studies with different patient populations, baseline demographics, outcomes, and study methods are also not ideal. For example, the JULIET and the TRANSCEND studies allowed for bridging therapy after apheresis and before CAR T-cell administration, whereas ZUMA-1 did not.17,28,35 Methodological differences and patient heterogeneity in the various clinical trials confound and obscure any safety, efficacy, or durability differences among the available second-generation CAR T-cell agents, precluding direct or indirect treatment comparisons. The National Comprehensive Cancer Network Clinical Practice Guidelines do not differentiate between axicabtagene ciloleucel, lisocabtagene maraleucel, and tisagenlecleucel in the third-line or subsequent line of therapy for most cases of DLBCL.2
Currently, randomized phase 3 clinical trials comparing the safety, efficacy, and tolerability of CAR T-cell therapy to standard-of-care second-line chemoimmunotherapy with autologous stem-cell transplantation in adults with LBCL are ongoing.44-46 These highly anticipated confirmatory clinical trials should help to refine patient selection criteria and determine the optimal sequencing of CAR T-cell therapies and autologous transplant in patients with select relapsed or refractory B-cell lymphomas.
Safety of CAR T-Cell Therapy
As “living drugs,” the use of CAR T-cell therapy results in markedly disparate adverse events compared with conventional chemotherapy.47,48 These unique complications may be life-threatening, and may warrant vigilant monitoring and expert navigation. The common adverse events identified in phase 1 and 2 clinical trials of CAR T-cell therapies include, but are not limited to, transient or persistent cytopenias, infection, cytokine-release syndrome (CRS), immune effector cell–associated neurotoxicity syndrome (ICANS), and macrophage activation syndrome or hemophagocytic lymphohistiocytosis.47,48
Additional adverse events include cross-reactivity with nontarget proteins, anaphylaxis, gastrointestinal disturbances, and tumor lysis syndrome. CRS and ICANS are the most notable adverse events associated with CAR T-cell therapy and present a significant barrier for the widespread adoption of the use of this treatment.47,48
In 2019, the American Society for Transplantation and Cellular Therapy (ASTCT) published consensus recommendations for defining and grading CRS and ICANS in patients receiving immune effector cell therapy.49 Early attempts to define and grade CRS and ICANS were incongruous across institutions and clinical trials, resulting in noteworthy practice and reporting differences that restricted the ability of clinicians and researchers to compare CAR T-cell agents and treatment schemes.49 In 2020, the Society for Immunotherapy of Cancer (SITC) convened an expert panel to provide clear, cohesive practice guidelines for the management of CRS and ICANS associated with CAR T-cell therapy.48
The creation and ongoing implementation of the ASTCT and the SITC guidelines can help to harmonize the definitions, grading criteria, and management strategies for CRS and ICANS for future clinical trials and commercial use of CAR T-cell therapy.
Cytokine-Release Syndrome
CRS is a common acute systemic inflammatory syndrome, typically manifesting within 1 to 2 weeks after CAR T-cell infusion.50 After recognizing and binding to the desired antigen (eg, CD19), CAR T-cells undergo in-vivo activation and proliferation. Many cytokines and chemokines—including interleukin (IL)-6, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, IL-1, IL-2, IL-2 receptor-α, IL-8, and IL-10—have been implicated in the generalized immune system activation leading to CRS.51 The pathophysiology of CRS is not fully understood; however, the recruitment and activation of bystander cells (ie, macrophages and monocytes) may initiate cytokine release and the hallmark proinflammatory response characteristic of CRS.
The hallmark clinical features of CRS include fever, hypotension, and hypoxia.50,52 Additional symptoms include arthralgias, headache, malaise, rigors, and tachycardia.50,52 CRS is often mild and reversible but may progress to a more severe syndrome, involving shock and multiorgan system failure that resembles macrophage activation syndrome or hemophagocytic lymphohistiocytosis.49
Macrophage activation syndrome and hemophagocytic lymphohistiocytosis are clinically overlapping life-threatening immune dysregulated processes mediated by overactive macrophages, hypercytokinemia, and hemophagocytosis.48 These hyperinflammatory syndromes result in clinical manifestations similar to CRS, including fever, elevated ferritin levels, and cytopenias. Fulminant macrophage activation syndrome and hemophagocytic lymphohistiocytosis are associated with mortality rates as high as 80%.48 The incidence of grade ≥3 CRS in B-cell lymphoma clinical trials ranged from 2% to 22%.17,28,35,38
CAR T-cell constructs with a CD28 co-stimulatory domain (eg, axicabtagene ciloleucel and brexucabtagene autoleucel) are associated with an increase in severe adverse events compared with those with 4-1BB.53 Overall, pivotal phase 2 clinical trials evaluating anti-CD19 CAR T-cell therapies in B-cell lymphomas reported a median time to CRS onset of between 2 and 5 days (range, 1-14 days), with a median duration of 7 to 11 days.17,28,35,38 A higher degree of disease burden before treatment with CAR T-cell therapy is associated with more toxicity, which is indicated by an increase in serum cytokines and CRS severity.47
Immune Effector Cell–Associated Neurotoxicity Syndrome
ICANS is another frequently occurring adverse event resulting from immune effector cell therapy.47 ICANS often occurs concomitantly or shortly after CRS; however, the 2 complications may occur separately.47 Although the exact underlying mechanism for ICANS is unknown, researchers have hypothesized that systemic inflammation, endothelial activation, and elevated cytokines can disrupt the blood–brain barrier permeability and result in aberrant levels of circulating cytokines in the cerebral spinal fluid.54 Similar to CRS, this process often involves IL-2, IL-6, IFN-γ, and TNF-α.55,56
The common manifestations of ICANS include aphasia, impaired attention, decreased level of consciousness, confusion, and motor impairment or weakness. The severe symptoms of ICANS include encephalopathy, seizures, and cerebral edema.49 The Immune Effector Cell–Associated Encephalopathy score can be used as a daily tool to assess frequently for ICANS and to trend neurologic progression.49 In addition, a baseline neurologic assessment should be performed before CAR T-cell therapy begins.48
The risk factors for ICANS include high disease burden, the use of a CD28-containing CAR T-cell therapy (eg, axicabtagene ciloleucel or brexucabtagene autoleucel), grade ≥3 CRS, receipt of high-intensity lymphodepleting chemotherapy, preexisting neurologic comorbidities, and younger age.48,57 The onset of ICANS can happen within the first 4 or 5 days and up to 1 month after infusion.28,50 The median time to the onset of ICANS after anti-CD19 CAR T-cell administration was 5 to 9 days (range, 1-66 days), with symptom resolution occurring after a median of 11 to 17 days.17,28,35,38
Hematologic Toxicity
CAR T-cell therapy may also result in hematologic adverse events, including cytopenias, coagulopathies, hypogammaglobulinemia, and long-standing B-cell aplasia.58 The etiology of these complications is often multifactorial as a result of conditioning chemotherapy before the infusion of CAR T-cells. In a study of 38 patients with relapsed or refractory B-cell ALL or lymphoma, early and late hematologic events were analyzed after treatment with CD28-containing anti-CD19 CAR T-cell therapies.58 After infusion with anti-CD19 CAR T-cell therapy, neutropenia occurred in 94% of patients, thrombocytopenia in 80%, and anemia in 51%. Overall, 93% of observed cytopenias occurred ≥21 days from cell infusion. Late-onset hematologic adverse events were identified more frequently in patients with recent stem-cell transplantation and high-grade CRS.58
Currently, CD19 is the safest and most effective antigen; however, noteworthy cytotoxicity occurs in malignant and healthy B-cells.12 CD19 is a frequently expressed B-cell marker, resulting in on-target, off-tumor complications. For example, B-cell aplasia has been reported as a possible long-term consequence of anti-CD19 CAR T-cell therapy.55,59 Hypogammaglobulinemia, which results from the destruction of normal B-cells, can occur as early as 2 to 3 months after CAR T-cell infusion and may persist for years after treatment.60 The subsequent decrease in antibody production predisposes patients to potentially life-threatening infections.60
Management of Adverse Events Associated with CAR T-Cell Therapy
All patients receiving CAR T-cell therapy must be diligently monitored for potential adverse events. Table 3 outlines the clinical manifestations associated with each CRS grade and the corresponding treatment recommendations. Acetaminophen and cooling blankets can be used to control temperature. Tocilizumab, a humanized antibody targeting the soluble IL-6 receptor, can be used for the treatment of grade ≥2 CRS.61 Tocilizumab was initially approved for the treatment of various arthritic conditions; however, because of its ability to interfere with the IL-6 signaling pathway, in 2017 tocilizumab was approved by the FDA for the treatment of CRS and is now the standard-of-care treatment for severe CRS.61
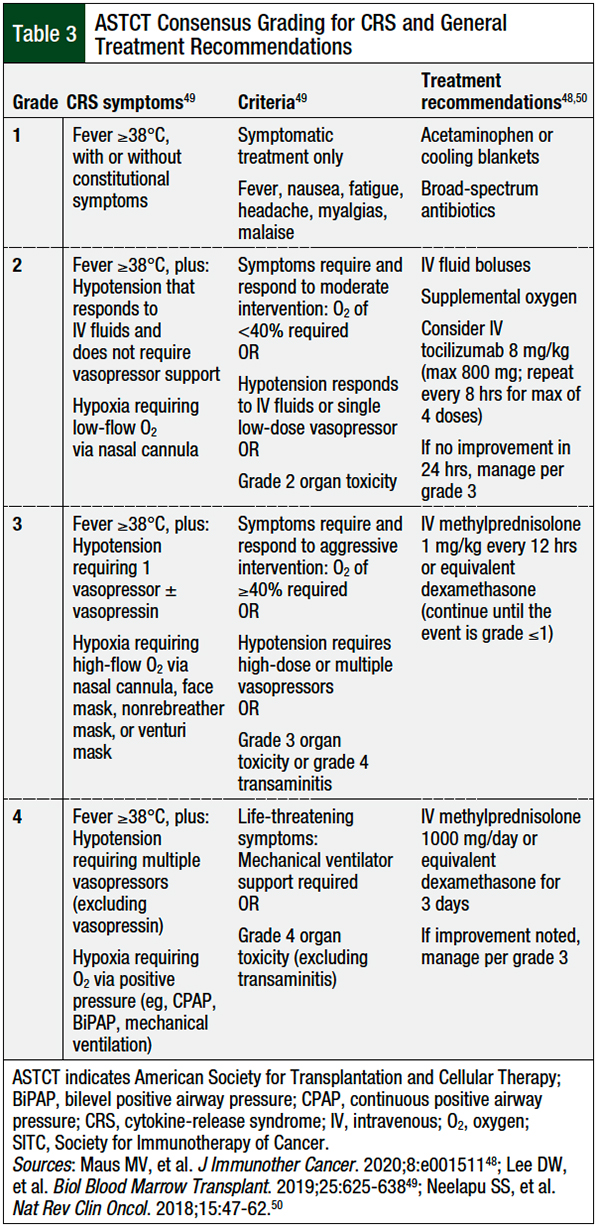
Several studies have evaluated nontraditional approaches to the initiation of tocilizumab treatment, with early intervention or the preemptive use of anti–IL-6 therapy resulting in a decreased incidence of severe CRS in high-risk patients.62-64 The use of tocilizumab does not negate the clinical efficacy of available CAR T-cell therapies and remains a key management strategy to minimize the progression of CRS.48,65
For patients with tocilizumab-refractory or severe CRS, the use of high-dose corticosteroids is recommended to suppress the inflammatory response, thereby mitigating the proinflammatory consequences of CRS.48 Although effective at reducing the inflammatory response in CRS, corticosteroids are lymphotoxic and may diminish the therapeutic effect of CAR T-cells. Further prospective studies are needed to optimize the dosing and timing of corticosteroids for patients with CRS and concurrent neurologic toxicities.
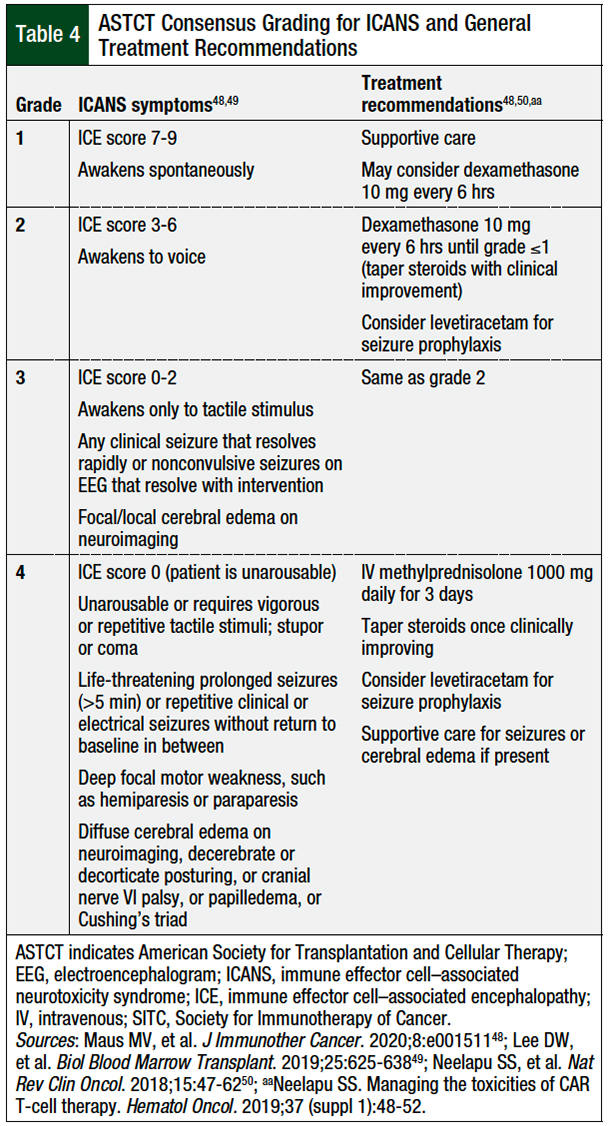
Unlike CRS, ICANS typically will not respond to IL-6 inhibition, because tocilizumab does not readily cross the blood–brain barrier.48,49 Furthermore, tocilizumab may even exacerbate ICANS by transiently elevating cerebral spinal fluid concentrations of IL-6.48,52 Therefore, corticosteroids and supportive care are included in the first-line therapy for ICANS.48
Table 4 details the grading criteria and treatment recommendations for ICANS.48,49 Because of the lymphotoxic effects of corticosteroids, clinicians should initiate a quick taper once the patient has shown improvement and clinical stabilization. Seizure prophylaxis, typically with levetiracetam, may be considered in high-risk patients.48
The non–FDA-approved immunosuppressants that are considered as treatments for tocilizumab-refractory CRS include siltuximab, an IL-6 inhibitor; anakinra, an IL-1 inhibitor; etanercept, a TNF inhibitor; sarilumab, a high-affinity IL-6 receptor antagonist; ibrutinib, a BTK inhibitor; and ruxolitinib, an oral Janus kinase/signal transducer and activator of transcription inhibitor.12,50,65-68
Unlike tocilizumab, which binds to the IL-6 receptor,61 siltuximab binds to circulating IL-6 directly, theoretically reducing ICANS risk.68 Other than corticosteroids, siltuximab is the most frequently used therapy as an alternative to tocilizumab for the treatment of severe and refractory CRS.69 Additional data are needed to further assess these salvage options in the treatment of severe CRS and ICANS.
Various strategies can be used to manage the hematologic adverse events associated with CAR T-cell therapy. Lymphodepleting conditioning regimens, B-cell aplasia, and hypogammaglobinemia place patients at risk for infectious complications, including bacterial, viral, fungal, and opportunistic infections.48 When determining infection prophylaxis, the SITC panel recommends that all patients receive pneumocystis pneumonia prophylaxis. Further consideration is warranted for prophylaxis against viral, fungal, and bacterial infections based on the patient’s previous lines of myelosuppressive therapy, infection history, prolonged steroid use, persistent neutropenia, anticytokine therapy, and high-dose lymphodepletion.48
Overall, infection precautions and prophylaxis are often institution-specific and aim to reduce the incidence and severity of complications. For patients with long-standing B-cell aplasia and subsequent hypogammaglobinemia, use of intravenous infusions of immunoglobulins, given every 4 to 6 weeks at 400 to 600 mg/kg should be considered to boost circulating antibodies to ≥400 mg/dL, until B-cell recovery and antibody formation.70
Risk Evaluation and Mitigation Strategy Program
Tisagenlecleucel, lisocabtagene maraleucel, axicabtagene ciloleucel, and brexucabtagene autoleucel have established FDA-mandated Risk Evaluation and Mitigation Strategy (REMS) programs to improve the management of CRS and ICANS.4,7-9 To administer CAR T-cell therapies, certified treatment centers must provide ongoing training for all personnel involved in the prescribing, dispensing, or administration of CAR T-cell therapy.
Because of the possibility of rapid clinical deterioration, REMS programs require treatment centers to always have 2 doses of tocilizumab immediately available for each patient receiving CAR T-cells.4,7-9 All CRS and ICANS events must be reported to the REMS program. In addition, patients may be required to live within close proximity to the treatment center for several weeks after treatment to ensure proper monitoring. Furthermore, each patient is required to have a wallet card identifying the drug they received and supplemental instructions.
Financial Concerns
CAR T-cell therapy carries financial risk for treatment centers and for patients.71,72 For the treatment of relapsed or refractory B-cell lymphoma, the average wholesale price for a single dose of a CAR T-cell agent ranges from $447,600 to $492,360.73 This range does not include any ancillary costs, such as for leukapheresis, hospitalization, possible intensive care unit care, tocilizumab therapy, early- and late-term monitoring, and other potential treatment requirements. Furthermore, payer reimbursement for CAR T-cell therapy continues to evolve and adapt as the commercial demand and consumption of immune effector cell therapy expand.72,74
Future Developments
Second-generation CAR-modified T-cell therapies have revolutionized the therapeutic landscape for various B-cell malignancies. Despite their early and ongoing successes, the current CAR T-cell therapies have well-known disadvantages requiring further improvement. Third-generation or later therapies are being evaluated to enhance T-cell cytotoxicity, expansion, and persistence, while also minimizing toxicity and resistance.75
Modifications to the CAR construct may augment antitumor response and enhance functional persistence.76 One such area of investigation includes genetic manipulation and further refinement of the specific co-stimulatory domains.76 Other investigational third-generation drugs include 2 co-stimulatory domains, such as CD28 plus 4-1BB, toll-like receptor 2, or OX40.77,78
Several CD19 antigen escape mechanisms have been identified, leading to disease recurrence and resistance to future anti-CD19 therapy.79 The emergence of CD19 antigen loss after anti-CD19 CAR T-cell therapy is an important mechanism necessitating additional research. Notable options to overcome the loss of CD19 target antigen include bispecific or trispecific CAR T-cell therapies, which aim to pair CD19-specific CARs with other frequently occurring B-cell antigens (eg, CD20) to avoid or overcome CD19 escape variants.80,81
Armored CAR T-cell therapies, also known as fourth-generation or next-generation CAR T-cell therapies, possess recalibrated co-stimulatory domains and are engineered to overcome tumor heterogeneity and a hostile tumor microenvironment.75 These armored CAR T-cell therapies either secrete peptides (eg, proinflammatory cytokines) or incorporate chemokines, or they switch membrane receptors to improve the tumor milieu and enhance T-cell activity.82,83
The CAR T-cell process is time- and resource-intensive, and is often inundated with significant logistical concerns. Patients enrolled in the phase 2 clinical trials discussed here had a median time from leukapheresis to infusion of 3 to 5 weeks, with 8% to 30% of enrolled patients not receiving CAR T-cell infusion after leukapheresis.17,28,35,38
To minimize these delays, interest in allogeneic so-called off-the-shelf CAR T-cells from healthy donors remains an intriguing option for future development.84 Universal off-the-shelf CAR T-cell therapies would provide a readily available treatment, without harvesting or manufacturing delays, while providing potential benefits, such as improving overall costs; alleviating lymphoma-cell contamination during apheresis; allowing for future redosing, if needed; and expanding access to innovative and potentially life-saving immunotherapy.84
For these and other reasons, allogeneic CAR T-cell therapies may provide another evolution in cancer treatment. Nevertheless, allogeneic CAR T-cell therapies also possess inherent drawbacks, such as the risk for life-threatening graft-versus-host disease and rapid elimination of the CAR T-cells by the host system, which must be considered.85
Conclusion
Genetically modified anti-CD19 CAR T-cell therapies are a paradigm-shifting therapeutic option in the treatment of advanced relapsed or refractory B-cell lymphomas. With unprecedented response rates providing deep and durable remissions in heavily pretreated patients, CAR T-cell therapy has evolved into a leading driver in the fight against cancer. However, autologous CAR T-cell therapies also elicit unique and life-threatening adverse events that require careful monitoring and expert management.
Propelled by the recent successes of second-generation CAR T-cell therapies and a strong desire to improve tolerability and logistic barriers, innovative third- and next-generation autologous or allogeneic drugs are in the drug pipeline. Randomized phase 3 clinical trials are needed to evaluate the use of CAR T-cell therapies according to the current standards of care in the second- or third-line treatment of patients with relapsed or refractory B-cell lymphoma.
Author Disclosure Statement
Dr Wells, Dr Summerlin, and Dr Halford have no conflicts of interest to report.
References
- Swerdlow SH, Harris NL, Jaffe ES, et al, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Rev 4th ed. Lyon, France: International Agency for Research on Cancer; 2017.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): B-Cell Lymphomas. Version 5.2021. September 22, 2021. www.nccn.org/professionals/physician_gls/pdf/b-cell.pdf. Accessed January 10, 2022.
- Coiffier B, Lepage E, Brière J, et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235-242.
- Kymriah (tisagenlecleucel) suspension for intravenous infusion [prescribing information]. Novartis Pharmaceuticals Corporation; August 2021. www.novartis.us/sites/www.novartis.us/files/kymriah.pdf. Accessed January 10, 2022.
- Kuwana Y, Asakura Y, Utsunomiya N, et al. Expression of chimeric receptor composed of immunoglobulin-derived V regions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960-968.
- Braendstrup P, Levine BL, Ruella M. The long road to the first FDA-approved gene therapy: chimeric antigen receptor T cells targeting CD19. Cytotherapy. 2020;22:57-69.
- Yescarta (axicabtagene ciloleucel) suspension for intravenous infusion [prescribing information]. Kite Pharma; April 2021. www.gilead.com/-/media/files/pdfs/medicines/oncology/yescarta/yescarta-pi.pdf. Accessed January 10, 2022.
- Tecartus (brexucabtagene autoleucel) suspension for intravenous infusion [prescribing information]. Kite Pharma; October 2021. www.gilead.com/-/media/files/pdfs/medicines/oncology/tecartus/tecartus-pi.pdf. Accessed January 10, 2022.
- Breyanzi (lisocabtagene maraleucel) suspension for intravenous infusion [prescribing information]. Juno Therapeutics, a Bristol-Myers Squibb Company; February 2021. Accessed January 10, 2022.
- Ehrlich P. Ueber den jetzigen Stand der Karzinomforschung. Ned Tijdschr Geneeskd. 1909;5:273-290.
- June CH, Maus MV, Plesa G, et al. Engineered T cells for cancer therapy. Cancer Immunol Immunother. 2014;63:969-975.
- Cerrano M, Ruella M, Perales MA, et al. The advent of CAR T-cell therapy for lymphoproliferative neoplasms: integrating research into clinical practice. Front Immunol. 2020;11:888. doi: 10.3389/fimmu.2020.00888.
- Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245-1256.
- Mueller KT, Waldron E, Grupp SA, et al. Clinical pharmacology of tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin Cancer Res. 2018;24:6175-6184.
- Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunology. 2019;8:e1049. doi: 10.1002/cti2.1049.
- Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015. doi: 10.1038/mto.2016.15.
- Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839-852.
- Kite. U.S. FDA approves Kite’s Tecartus, the first and only CAR T treatment for relapsed or refractory mantle cell lymphoma. July 24, 2020. www.kitepharma.com/news/press-releases/2020/7/us-fda-approves-kites-tecartus-the-first-and-only-car-t-treatment-for-relapsed-or-refractory-mantle-cell-lymphoma. Accessed May 13, 2021.
- Klebanoff CA, Khong HT, Antony PA, et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111-117. Erratum in: Trends Immunol. 2005;26:298.
- Riedell PA, Bishop MR. Safety and efficacy of axicabtagene ciloleucel in refractory large B-cell lymphomas. Ther Adv Hematol. 2020;11:2040620720902899. doi: 10.1177/2040620720902899.
- Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720-724.
- Rosenberg SA, Aebersold P, Cornetta K, et al. Gene transfer into humans—immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570-578.
- Goverman J, Gomez SM, Segesman KD, et al. Chimeric immunoglobulin-T cell receptor proteins form functional receptors: implications for T cell receptor complex formation and activation. Cell. 1990;60:929-939.
- Hwu P, Yang JC, Cowherd R, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55:3369-3373.
- Cooper LJN, Topp MS, Serrano LM, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B–lineage leukemia effect. Blood. 2003;101:1637-1644.
- Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509-1518. Erratum in: N Engl J Med. 2016;374:998.
- Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099-4102.
- Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531-2544.
- US Food and Drug Administration. FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma. October 18, 2017. www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma. Accessed December 20, 2021.
- Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31-42.
- Jacobson C, Locke FL, Ghobadi A, et al. Long-term (4- and 5-year) overall survival in ZUMA-1, the pivotal study of axicabtagene ciloleucel (axi-cel) in patients with refractory large B-cell lymphoma (LBCL). Poster presented at the American Society of Hematology annual meeting; December 11-14, 2021; Atlanta, GA. Abstract 1764.
- Jacobson C, Chavez JC, Sehgal AR, et al. Primary analysis of Zuma-5: a phase 2 study of axicabtagene ciloleucel (axi-cel) in patients with relapsed/refractory (R/R) indolent non-Hodgkin lymphoma (iNHL). Blood. 2020;136(suppl 1):40-41.
- US Food and Drug Administration. FDA grants accelerated approval to axicabtagene ciloleucel for relapsed or refractory follicular lymphoma. March 5, 2021. www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-axicabtagene-ciloleucel-relapsed-or-refractory-follicular-lymphoma. Accessed August 9, 2021.
- US Food and Drug Administration. FDA approval brings first gene therapy to the United States. August 30, 2017. www.fda.gov/news-events/press-announcements/fda-approval-brings-first-gene-therapy-united-states. Accessed May 20, 2021.
- Schuster SJ, Bishop MR, Tam CS, et al; for the JULIET investigators. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380:45-56.
- US Food and Drug Administration. FDA approves tisagenlecleucel for adults with relapsed or refractory large B-cell lymphoma. May 3, 2018. www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-adults-relapsed-or-refractory-large-b-cell-lymphoma. Accessed December 21, 2021.
- Chong EA, Ruella M, Schuster SJ; for the Lymphoma Program Investigators at the University of Pennsylvania. Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N Engl J Med. 2021;384:673-674.
- Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382:1331-1342.
- Jain P, Wang M. Mantle cell lymphoma: 2019 update on the diagnosis, pathogenesis, prognostication, and management. Am J Hematol. 2019;94:710-725.
- US Food and Drug Administration. FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma. July 24, 2020. www.fda.gov/drugs/fda-approves-brexucabtagene-autoleucel-relapsed-or-refractory-mantle-cell-lymphoma. Accessed August 8, 2021.
- Wang M, Munoz J, Goy AH, et al. One-year follow-up of ZUMA-2, the multicenter, registrational study of KTE-X19 in patients with relapsed/refractory mantle cell lymphoma. Blood. 2020;136(suppl 1):20-22.
- US Food and Drug Administration. FDA approves lisocabtagene maraleucel for relapsed or refractory large B-cell lymphoma. February 5, 2021. www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-lisocabtagene-maraleucel-relapsed-or-refractory-large-b-cell-lymphoma. Accessed August 8, 2021.
- Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130:1800-1808. Erratum in: Blood. 2018;131:587-588.
- ClinicalTrials.gov. Tisagenlecleucel in adult patients with aggressive b-cell non-Hodgkin lymphoma (BELINDA). ClinicalTrials.gov identifier NCT03570892. Updated November 8, 2021. https://clinicaltrials.gov/ct2/show/NCT03570892. Accessed December 22, 2021.
- ClinicalTrials.gov. Efficacy of axicabtagene ciloleucel compared to standard of care therapy in subjects with relapsed/refractory diffuse large B cell lymphoma (ZUMA-7). ClinicalTrials.gov identifier NCT03391466. Updated October 14, 2021. https://clinicaltrials.gov/ct2/show/NCT03391466. Accessed December 22, 2021.
- ClinicalTrials.gov. A study to compare the efficacy and safety of JCAR017 to standard of care in adult subjects with high-risk, transplant-eligible relapsed or refractory aggressive B-cell non-Hodgkin lymphomas (TRANSFORM). ClinicalTrials.gov identifier NCT03575351. Updated June 10, 2021. https://clinicaltrials.gov/ct2/show/NCT03575351. Accessed December 22, 2021.
- Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321-3330.
- Maus MV, Alexander S, Bishop MR, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. J Immunother Cancer. 2020;8:e001511. doi: 10.1136/jitc-2020-001511.
- Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25:625-638.
- Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15:47-62.
- Teachey DT, Lacey SF, Shaw PA, et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016;6:664-679.
- Chen H, Wang F, Zhang P, et al. Management of cytokine release syndrome related to CAR-T cell therapy. Front Med. 2019;13:610-617.
- Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021;56:552-566.
- Gust J, Hay KA, Hanafi LA, et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7:1404-1419.
- Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507-1517. Erratum in: N Engl J Med. 2016;374:998.
- Titov A, Petukhov A, Staliarova A, et al. The biological basis and clinical symptoms of CAR-T therapy-associated toxicities. Cell Death Dis. 2018;9:897. doi: 10.1038/s41419-018-0918-x.
- Karschnia P, Jordan JT, Forst DA, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood. 2019;133:2212-2221.
- Fried S, Avigdor A, Bielorai B, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019;54:1643-1650.
- Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 2012;119:2709-2720.
- Doan A, Pulsipher MA. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr Blood Cancer . 2018;65:e26914. doi: 10.1002/pbc.26914.
- Actemra (tocilizumab) injection, for intravenous or subcutaneous use [prescribing information]. Genentech; March 2021. www.gene.com/download/pdf/actemra_prescribing.pdf. Accessed January 10, 2022.
- Locke FL, Neelapu SS, Bartlett NL, et al. Preliminary results of prophylactic tocilizumab after axicabtagene ciloleucel (axi-cel; KTE-C19) treatment for patients with refractory, aggressive non-Hodgkin lymphoma (NHL). Blood. 2017;130(suppl 1):1547.
- Gardner RA, Ceppi F, Rivers J, et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood. 2019;134:2149-2158.
- Kadauke S, Myers RM, Li Y, et al. Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial. J Clin Oncol. 2021;39:920-930.
- Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188-195. Errata in: Blood. 2015;126:1048; Blood. 2016;128:1533.
- Kevzara (sarilumab) injection, for subcutaneous use [prescribing information]. Regeneron Pharmaceuticals/sanofi-aventis US; April 2018. https://products.sanofi.us/Kevzara/Kevzara.pdf. Accessed January 10, 2022.
- Kenderian SS, Ruella M, Shestova O, et al. Ruxolitinib prevents cytokine release syndrome after Car T-cell therapy without impairing the anti-tumor effect in a xenograft model. Biol Blood Marrow Transplant. 2017;23:S19-S20.
- van Rhee F, Fayad L, Voorhees P, et al. Siltuximab, a novel anti–interleukin-6 monoclonal antibody, for Castleman’s disease. J Clin Oncol. 2010;28:3701-3708.
- Mahmoudjafari Z, Hawks KG, Hsieh AA, et al. American Society for Blood and Marrow Transplantation Pharmacy Special Interest Group survey on chimeric antigen receptor T cell therapy administrative, logistic, and toxicity management practices in the United States. Biol Blood Marrow Transplant. 2019;25:26-33.
- Chakraborty R, Hill BT, Majeed A, Majhail NS. Late effects after chimeric antigen receptor T cell therapy for lymphoid malignancies. Transplant Cell Ther. 2021;27:222-229.
- Prasad V. Tisagenlecleucel—the first approved CAR-T-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol. 2018;15:11-12.
- Manz CR, Porter DL, Bekelman JE. Innovation and access at the mercy of payment policy: the future of chimeric antigen receptor therapies. J Clin Oncol. 2020;38:384-387.
- IBM Watson Health/Truven Health Analytics. IBM Micromedex Web Applications Access. www.micromedexsolutions.com [requires subscription]. Accessed January 18, 2022.
- Bessette Z. CMS hospital IPPS proposed rule includes CAR-T reimbursement updates. J Clin Pathw. 2020;6:14.
- Huang R, Li X, He Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13:86. doi: 10.1186/s13045-020-00910-5.
- Feucht J, Sun J, Eyquem J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25:82-88. Erratum in: Nat Med. 2019;25:530.
- George P, Dasyam N, Giunti G, et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: a phase I clinical trial protocol (ENABLE). BMJ Open. 2020;10:e034629. doi: 10.1136/bmjopen-2019-034629.
- Park JH, Brentjens RJ. Adoptive immunotherapy for B-cell malignancies with autologous chimeric antigen receptor modified tumor targeted T cells. Discov Med. 2010;9:277-288.
- Jacobson CA, Hunter B, Armand P, et al. Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance and toxicity. Blood. 2018;132(suppl 1):92.
- Zah E, Lin MY, Silva-Benedict A, et al. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4:498-508. Addendum in: Cancer Immunol Res. 2016;4:639-641.
- Fousek K, Watanabe J, Joseph SK, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35:75-89.
- Liu Y, Di S, Shi B, et al. Armored inducible expression of IL-12 enhances antitumor activity of glypican-3–targeted chimeric antigen receptor–engineered T cells in hepatocellular carcinoma. J Immunol. 2019;203:198-207.
- Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15:1145-1154.
- Depil S, Duchateau P, Grupp SA, et al. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185-199.
- Aftab BT, Sasu B, Krishnamurthy J, et al. Toward “off-the-shelf” allogeneic CAR T cells. Adv Cell Gene Ther. 2020;3:e86. doi: 10.1002/acg2.86.