This section provides a summary of symptomatic conditions in oncology and their management. Readers are invited to submit summaries following the guidelines.
Justin Arnall, PharmD, BCOP, Clinical Pharmacist, Hematology, Atrium Health Specialty Pharmacy Service, Charlotte, NC; Joseph Elmes, PharmD, BCOP, Clinical Staff Pharmacist, Atrium Health Levine Cancer Institute, Charlotte, NC; Umesh Yogarajah, PharmD, Clinical Specialist, Malignant Hematology, Roswell Park Comprehensive Cancer Center, Buffalo, NY; Benson Meek, PharmD, Staff Pharmacist, Atrium Health Specialty Pharmacy Service, Charlotte, NC; Donald C. Moore, PharmD, BCPS, BCOP, DLPA, FCCP, Clinical Oncology Pharmacy Manager, Atrium Health Levine Cancer Institute, Charlotte, NC.
Corresponding author: Justin Arnall, jarnall87@gmail.com
Connecting Science to Practice
Ruxolitinib discontinuation syndrome can be a potential
life-threatening complication associated with the
abrupt discontinuation of ruxolitinib therapy when
used for the treatment of myelofibrosis. Key risk factors
associated with ruxolitinib discontinuation syndrome
include myelofibrosis disease severity, patient frailty,
and hematologic parameters. Mitigating this adverse
event can be accomplished by tapering ruxolitinib
doses gradually and avoiding abrupt discontinuation.
In addition, prophylactic corticosteroids can be considered
while tapering ruxolitinib to further aid in preventing
this syndrome. It is not well-elucidated if other
JAK inhibitors recently approved for the treatment of
myelofibrosis also carry a significant risk for discontinuation
syndrome.
This article focuses on elucidating the significance of ruxolitinib discontinuation syndrome (RDS) in patients with myelofibrosis, particularly in addressing its symptoms, etiology, treatment options, and strategies for mitigating its occurrence during treatment discontinuation and transitions between JAK inhibitors. This review underscores the critical implications of RDS among hematology and oncology pharmacists, offering vital insights into managing this severe adverse event (AE).
Symptom Overview
Ruxolitinib was the first approved JAK inhibitor for the treatment of myelofibrosis. Since its approval and widespread use for this indication, rare events of a withdrawal or discontinuation syndrome around the time of therapy discontinuation, occurring between 2 and 21 days, have been described and subsequently evaluated.1 RDS is a cytokine release–type syndrome that is characterized by a sudden relapse of symptoms and splenomegaly on therapy discontinuation and presents as a rare yet serious AE, with severe manifestations leading to life-threatening conditions and even death.1
Although standardized protocols for the identification and management of RDS are still lacking, the identified risk factors for RDS include disease severity, patient frailty, and certain hematologic parameters.1 The lack of established guidelines necessitates a comprehensive understanding of potential prevention strategies for RDS, including tapering approaches, prophylactic corticosteroids, and close monitoring on discontinuation. Further, based on the kinetic and dynamic characteristics of recently approved novel JAK inhibitors (fedratinib, pacritinib, momelotinib), there may be differences in the risk for RDS. The treatment options for RDS primarily involve restarting treatment with JAK inhibitors, supportive measures, and possibly corticosteroid administration.1 Insight into the application of these strategies regarding the novel JAK inhibitors is still limited, but strategies may be developed based on clinical trial data.
Therefore, there is a need for standardized approaches to the management of RDS during transitions between JAK inhibitors, which is especially important to consider in an era of novel JAK inhibitors. Clinicians should use the available insights on RDS prevention and management and tailor treatment strategies and interventions to individual patient scenarios, while also seeking optimal standard approaches to care to aim for improved patient outcomes and minimized AEs in the treatment of myelofibrosis.
Myelofibrosis is characterized by ineffective hepatosplenic, extramedullary hematopoiesis that results from fibrotic changes to the bone marrow.2 This cancer is a myeloproliferative neoplasm and includes primary myelofibrosis as well as postessential thrombocythemia and post–polycythemia vera myelofibrosis.2 Myelofibrosis is associated with systemic manifestations that result from the abnormal release of cytokines.2 Mutations in the JAK family, which play a crucial role in signal transduction for growth factors and cytokines, are frequently observed in patients with myelofibrosis.2 Ruxolitinib was the first JAK inhibitor approved for the treatment of myelofibrosis to reduce splenomegaly and alleviate disease-related symptoms.1 Despite remarkable efficacy, many patients do not reach or maintain a response or are intolerant of treatment with ruxolitinib. Nonresponse after treatment with ruxolitinib is being studied with special interest now that additional JAK inhibitors have been approved for myelofibrosis, but clinicians expect approximately 40% of patients to discontinue ruxolitinib treatment within 3 years of therapy.1
In early clinical trials of ruxolitinib in patients with myelofibrosis, most patients had an acute relapse of their symptoms and accelerated splenomegaly after discontinuing treatment with ruxolitinib.3 In one of these trials, 5 of 47 symptomatic patients had life-threatening AEs, including respiratory distress, septic shock-like syndrome, and disseminated intravascular coagulation that required hospitalization.3 Several other cases have had a similar presentation after discontinuing treatment with ruxolitinib since then, suggesting that this RDS may be a rare but serious AE that can even be associated with death.4-7
A survey was recently conducted among a clinical network that sought to characterize ruxolitinib and subsequent outcomes among patients with myelofibrosis who received treatment with ruxolitinib at one of 22 participating institutions.1 The discontinuation symptoms included all new symptoms occurring within 21 days from the discontinuation of treatment with ruxolitinib and were interpreted by the treating hematologist as related to the discontinuation. A lack or loss of response or leukemic transformation was the main cause for stopping treatment with ruxolitinib, with AEs (mostly hematologic) as the secondary reason. Among the 251 patients who discontinued receiving ruxolitinib, 34 (13.5%) patients had RDS that was identified by their treating hematologist. The symptoms were mild in 21 (61.8%) patients, with noted splenomegaly and constitutional symptoms. A total of 10 (29.4%) patients had moderate symptoms requiring treatment with corticosteroids or the reinitiation of JAK inhibition. A total of 3 (8.8%) patients had severe symptoms, including spleen rupture, acute respiratory distress syndrome, and septic-like shock that required intensive care. The median time to symptoms was 7 days (range, 2-21 days) from the time that the treatment with ruxolitinib was stopped, although the cases of severe manifestations occurred within 48 hours from the discontinuation of treatment with ruxolitinib and then rapidly improved when ruxolitinib was rechallenged.1
Although the characterization of RDS is very useful in predicting such AEs regarding the discontinuation of ruxolitinib, insight was limited in identifying the best strategies for avoiding RDS.1 In this survey, a minority of institutions reported the tapering of ruxolitinib or using prophylactic corticosteroids. The identified risk factors for RDS include intermediate-2 risk and high-risk disease, patient frailty, transfusion dependency, platelet counts <100,000/mcL, unfavorable karyotype, and increasing peripheral blasts, however a standard for identification, avoidance, and management has yet to be established.5 Further, the approval of novel JAK inhibitors for myelofibrosis (fedratinib, pacritinib, and momelotinib) in the past few years prompts questions about the risk for discontinuation syndrome with these medications and when switching from one therapy to another. For this reason, the standardization and implementation of discontinuation strategies should be pursued. Whether stopping or switching JAK inhibitors, clinicians should consider approaches to minimize the risk for RDS along with careful monitoring and management plans.
Etiology
JAKs are cytoplasmic protein tyrosine kinases that are essential for signal transduction of inflammatory cytokines, including interleukin-6 and tumor necrosis factor-alpha, especially through the JAK-STAT signal pathway.3 STAT activation induces cytokines and growth factors, with most immune responses dependent on this pathway.4 The inhibition of JAK1 by ruxolitinib as an ATP mimetic reduces the production of several inflammatory cytokines as part of its activity to treat splenomegaly and ameliorate disease-related symptoms.1,8 Cytokine inhibition is critical here because abnormal expression contributes to myelofibrosis-related bone marrow stromal changes, ineffective erythropoiesis, extramedullary hematopoiesis, and constitutional symptoms.8,9 Despite being a cornerstone of therapy and remarkably improving outcomes in many patients, as many as 40% of patients in clinical trials did not achieve a stable response or had disease that was intolerant to treatment with ruxolitinib.5 In patients who required the discontinuation or dose reduction of ruxolitinib, an accumulation of JAK activation loop phosphorylation has been observed.5,8 Thus, RDS likely is caused by a hypersensitivity of suddenly reactivated JAK1 (and JAK2), which results in a cytokine storm and systemic inflammatory response.10
RDS is often a diagnosis of exclusion, although without specific diagnostic criteria, and early suspicion of RDS by clinicians may be crucial for improved outcomes.5 Sudden relapse of the patient’s symptoms with an accelerated blast crisis that leads to tumor lysis syndrome, worsening cytopenias, and increasing splenomegaly is the most frequent indication of RDS.11 Additional symptoms that have been reported with this discontinuation syndrome include fatigue, itching, bone pain, and constitutional symptoms.2 Several other severe AEs can occur with RDS, including acute respiratory distress syndrome, splenic infarction, shock-like syndrome, disseminated intravascular coagulopathy, and hypocalcemia.2 These events are likely associated with an acute rebound of cytokine production.2
Treatment Options
There are no standard guidelines for the prevention of RDS or standard recommendations for the management of its manifestations. To avoid the continued and serious complications of RDS symptoms, clinical trial investigators have recommended careful tapering under supervision since the identification of this complication, which reflects common practice.5,8 Avoiding the sudden discontinuation of ruxolitinib is a primary measure that can be used to prevent the occurrence of discontinuation syndrome. Prescribers may seek to taper doses by 5 to 10 mg daily over a 14-day period to a target of at most 10 mg twice daily (if not to a lower dose), especially in patients who have a high risk for rebound symptoms (high disease burden or refractory disease) or in patients who are deemed frail (Table). This may be difficult depending on the current dosing and tablet size and may require a new prescription to achieve. Administering prophylactic corticosteroids either during a taper or for a period of time after discontinuation has also been reported in real-world practice.1,5 Stopping ruxolitinib soon before the start of conditioning for allogeneic hematopoietic stem-cell transplantation may also prevent RDS.5
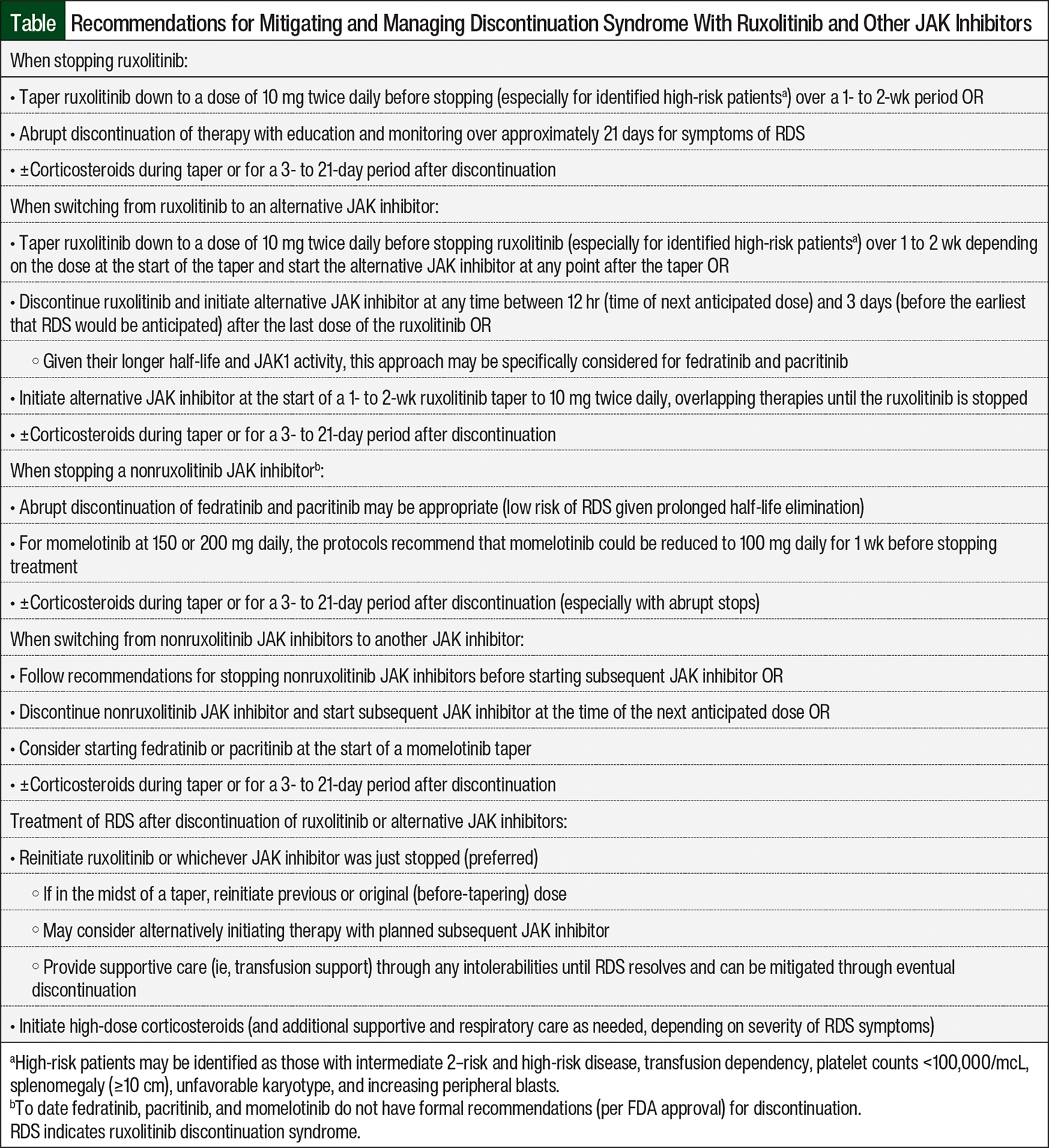
When RDS occurs, patients should be restarted on ruxolitinib if possible and supportive measures should be applied appropriately (Table). Supportive therapy may include red blood cell and platelet transfusions for severe cytopenias, the correction of electrolyte imbalances as a result of tumor lysis syndrome, plasma for disseminated intravascular coagulation, and high-dose systemic corticosteroids to reduce the cytokine storm.6 Interventions such as ventilator support may be needed in severe respiratory distress.6 The addition of corticosteroids may be considered even in the setting of mild-to-moderate symptoms if not already given prophylactically (Table). No specific recommendations for corticosteroid dosing are currently available, therefore institutions should consider and report outcomes on standardized approaches to therapy. In severe cases of RDS, high doses of corticosteroids can be considered and prolonged tapers may be used depending on symptom response.
There may be emergent situations where the sudden discontinuation of treatment with ruxolitinib is unavoidable. This may result from the need to mitigate severe cytopenias among other AEs, or because patients are hospitalized without the medication on hand and are unable to easily bring it in to continue therapy. Such situations resulting in particularly severe manifestations of RDS are reported in the literature as cases providing insight into presentation and management.3,4 To effectively manage this situation, health systems should consider establishing a standard of early inclusion of hematology and oncology specialists in the care of admitted patients who are receiving JAK inhibitors (specifically ruxolitinib) at home. Because patients who are hospitalized may initially present to nononcology units, internal medicine and critical care specialists may need to be aware of the risk for this withdrawal syndrome. Medication reconciliation will be critical in identifying this medication during transitions of care, which can avoid an unintended hold or discontinuation of ruxolitinib. For those patients who are unable to bring in their at-home medication, consider models of collaboration with a specialty pharmacy that may streamline dispensing the agent, or consider pursuing nonformulary approval with inpatient pharmacies (if ruxolitinib is not routinely available) to acquire medication in urgent situations.12 The ability to continue treatment with ruxolitinib, or even restart the drug if necessary, may be critical in avoiding severe disease manifestations.
Discontinuation syndrome has recently become an area of consideration with the approvals of alternative JAK inhibitors after ruxolitinib. First, there have been concerns regarding a similar syndrome with the other JAK inhibitors as with ruxolitinib, but with limited reports to date, which are thought to be in part because of longer half-lives, at least with fedratinib (41 hours) and pacritinib (27.7 hours).13 One case of discontinuation syndrome with pacritinib has been reported to date.13 After a short taper at 50% of the original dose of pacritinib over 1 week with subsequent discontinuation, the patient in this case had new-onset symptoms and clinical manifestations that are a hallmark of RDS. The time to onset was approximately 2 weeks after the discontinuation of treatment with pacritinib, which is a time to onset and natural history like RDS.13 Therefore, currently no tapering is formally recommended for these agents at treatment discontinuation.
Second, there is a lack of clear guidance on the optimal method for transitioning from treatment with ruxolitinib to alternative JAK inhibitors. Fedratinib and pacritinib have been evaluated in patients after treatment with ruxolitinib.8,14-16 Before switching from ruxolitinib to an alternative JAK inhibitor, there are several factors to consider regarding this syndrome, including the current dose, duration of therapy, reason for switching therapy (disease progression vs treatment intolerance), and patient-specific factors.16 Expert commentary suggests tapering ruxolitinib slowly over 14 days (while obtaining the alternative JAK inhibitor) and starting the alternative once ruxolitinib is completely stopped, switching directly from ruxolitinib to the other JAK inhibitor, and overlapping the 2 JAK inhibitors while tapering ruxolitinib (Table).1,9,14-16 The ultimate strategy will likely depend on the patient- and disease-specific scenarios, but there are limited data that can offer insight into the optimal switching strategy.
Fedratinib, pacritinib, and momelotinib were approved for use after the results of clinical trials that included patients who previously received ruxolitinib.17-23 With fedratinib, the JAKARTA-2 trial included patients with myelofibrosis who had received ruxolitinib therapy for at least 14 days (unless the patient had discontinued early as a result of intolerance or allergy).17 In addition, the study’s main exclusion criterion was receiving chemotherapy, including ruxolitinib, within 14 days before the start of the study (an exception was made for hydroxyurea, which was permitted within 1 day of starting treatment with fedratinib).17,18 There was also no recommendation for the tapering of fedratinib, because with the specificity of fedratinib for JAK2 inhibition, its weaker inhibition of JAK1 may offer explanation for the lack of concern for discontinuation syndrome.9 With pacritinib, the PERSIST-2 trial included patients who previously received ruxolitinib and switched treatment as a result of treatment intolerance or disease progression.19 Unfortunately, this trial did not offer insight into the switch from ruxolitinib to pacritinib.19 Subsequent to the PERSIST studies and in response to the clinical hold of pacritinib from the FDA, the PAC203 study was conducted to confirm the safety and optimal dosing of pacritinib, and included patients who did not previously respond to ruxolitinib therapy, which was defined as treatment for at least 3 months, with <10% spleen volume reduction or <30% decrease in spleen length or regrowth of the spleen.20 The exclusion criteria in PAC203 include patients receiving a high dose of ruxolitinib (>20 mg daily) who could not tolerate tapering to ≤10 mg twice daily for 14 days before the first dose of pacritinib.20 Given the longer half-life and JAK1-sparing coverage of fedratinib and pacritinib, a slow taper of ruxolitinib and possible overlap of therapies have been suggested, although there are little data in this approach to switching therapies.16
Momelotinib was approved for the treatment of myelofibrosis based on the results of the phase 3 SIMPLIFY-1, SIMPLIFY-2, and MOMENTUM studies.21-23 From a pharmacokinetic standpoint, the half-life elimination of momelotinib is short and similar to that of ruxolitinib compared with the significantly longer half-lives of fedratinib and pacritinib, which may suggest a risk for discontinuation syndrome similar to that of ruxolitinib.24,25 In SIMPLIFY-2 and MOMENTUM, patients were started on treatment with momelotinib after a loss of response or intolerability to earlier JAK inhibitor therapy (ruxolitinib), with a majority of patients transitioning from ruxolitinib.22,23
Regarding the discontinuation of momelotinib, both SIMPLIFY trials allowed tapering at treatment discontinuation or interruptions at the investigator’s discretion.21,22 For patients who were receiving the 150- or 200-mg daily dose, the protocols recommended that momelotinib could be reduced to 100 mg daily for 1 week before stopping treatment (and could be stopped immediately when at a dose of 100 mg daily).21,22 In SIMPLIFY-1, 27 of 85 (31.8%) patients receiving momelotinib who did not have their treatment tapered and 91 of 326 (27.9%) patients whose treatment was tapered had at least 1 AE.26 Furthermore, 10 of 85 (11.8%) patients whose treatment was not tapered and 55 of 326 (16.9%) patients whose treatment was tapered had ≥1 grade 3 or 4 AE. The most common grade 3/4 AEs were anemia (without taper, 2.4%; with taper, 2.8%) and thrombocytopenia (without taper, 3.5%; with taper, 2.5%). There were no cases of cytokine release syndrome–disseminated intravascular coagulation, respiratory distress, or multiorgan failure. There were 2 cases of hypotension and 2 cases of tachycardia in the patients whose treatment was tapered.21
In SIMPLIFY-2, 16 of 32 (50%) patients who did not receive tapered momelotinib and 42 of 112 (37.5%) patients who received tapered momelotinib had ≥1 AE.26 Furthermore, 7 of 32 (21.9%) patients who did not taper momelotinib and 30 of 112 (26.8%) patients who received tapered momelotinib had ≥1 grade 3/4 AE. There were no cases of cytokine release syndrome, disseminated intravascular coagulation, or multiorgan failure.26 There were 2 cases of respiratory distress (1 in each group), and there was 1 case of tachycardia in a patient who received tapered momelotinib.26 The MOMENTUM trial also allowed similar tapering, with the consideration of an additional incremental reduction of momelotinib to 50 mg daily (not commercially available).23 To date, there are no formal recommendations for the tapering of momelotinib, but this agent may be associated with a higher degree of risk and patients may be closely monitored regarding treatment discontinuation.
What we can glean from these clinical trial data is that the preferred practice may be to taper ruxolitinib to at least 10 mg twice daily, if not tapering off completely, before switching to an alternative JAK inhibitor at the next dosing time.13 It may be reasonable, and even preferred at higher doses, to stop treatment with ruxolitinib and start the alternative JAK treatment within 3 to 7 days, while educating the patient on recognizing the symptoms of discontinuation syndrome and having a plan ready to address. Patients may be able to best adhere to this plan of care. Including a short course of prednisone with the transition treatment may be considered as well.14,16 To date, overlapping JAK inhibitors (potentially while tapering treatment with ruxolitinib) seems the least supported and would not be recommended when there is concern for overlapping AEs. Overall, the approach to transitioning between ruxolitinib (as summarized in the Table) and other JAK inhibitors may vary depending on patient-specific situations, but clinical trial data and drug properties can be used to guide the strategies for doing so.
Conclusion
RDS has become an area of renewed consideration in recent years with the approval of multiple JAK inhibitors for the treatment of myelofibrosis. There have been concerns regarding the syndrome during transition between treatments, and clinical trials have included a washout period of ruxolitinib before starting another agent. In clinical practice and given the expectation of the syndrome occurring at a median of 7 days, most patients will switch directly from one drug to another without the complete washout of the first drug, but a variety of treatment-switching strategies may be considered.
With the effective management and reintroduction of JAK inhibition, the symptoms of RDS may be expected to resolve quickly within 24 to 48 hours; however, in the most severe cases of RDS or when the syndrome is not recognized quickly, patients’ symptoms may worsen and life-threatening or fatal outcomes may occur.
Author Disclosures
Dr Arnall is on the Advisory Board at CTI Biopharma and HEMA Pharmaceuticals and is on the Speaker’s Bureau at Novo Nordisk; Dr Moore is on the Advisory Board at Genentech, GSK, Incyte, and Sanofi and reports consulting with Genmab.
References
- Palandri F, Palumbo GA, Elli EM, et al. Ruxolitinib discontinuation syndrome: incidence, risk factors, and management in 251 patients with myelofibrosis. Blood Cancer J. 2021;11:4.
- Tefferi A. Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023;98:801-821.
- Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011;86:1188-1191.
- Houthuys JF, Wilmer AP, Peetermans M, et al. Severe ARDS due to ruxolitinib discontinuation syndrome: case presentation and literature review. Heliyon. 2022;8:e11782.
- Baek DW, Cho HJ, Lee JM, et al. Light and shade of ruxolitinib: positive role of early treatment with ruxolitinib and ruxolitinib withdrawal syndrome in patients with myelofibrosis. Expert Rev Hematol. 2022;15:573-581.
- Chukwuka NH, Hashmi AT, Kamholz SL. Ruxolitinib discontinuation syndrome. Am J Ther. 2020;29:e132-e134.
- Szpytma MM, Gimpel D, Crouch G, Bennetts JS. Ruxolitinib withdrawal complicating emergency aortic root replacement. Interact Cardiovasc Thorac Surg. 2022;35:ivac143.
- Tvorogov D, Thomas D, Liau NPD, et al. Accumulation of JAK activation loop phosphorylation is linked to type I JAK inhibitor withdrawal syndrome in myelofibrosis. Sci Adv. 2018;4:eaat3834.
- Talpaz M, Kiladjian JJ. Fedratinib, a newly approved treatment for patients with myeloproliferative neoplasm-associated myelofibrosis. Leukemia. 2021;35:1-17.
- Shakir AT, Nusrat S, Keruakous A. Ruxolitinib discontinuation syndrome in a patient with myelofibrosis to acute myeloid leukemia transformation. JCO Oncol Pract. 2020;16:395-396.
- Tefferi A. JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood. 2012;119:2721-2730.
- Wyatt H, Zuckerman AD, Hughes ME, et al. Addressing the challenges of novel oncology and hematology treatments across sites of care: specialty pharmacy solutions. J Oncol Pharm Pract. 2022;28:627-634.
- Handa S, Farina KA, Becker M, et al. Discontinuation syndrome with JAK2 selective agents: case presentation and mechanistic insights. JCO Precis Oncol. 2024;8:e2300234.
- England JT, Gupta V. Fedratinib: a pharmacotherapeutic option for JAK-inhibitor naive and exposed patients with myelofibrosis. Expert Opin Pharmacother. 2022;23:1677-1686.
- Mascarenhas JO, Verstovsek S. The clinical dilemma of JAK inhibitor failure in myelofibrosis: predictive characteristics and outcomes. Cancer. 2022;128:2717-2727.
- Bose P, Kuykendall AT, Miller C, et al. Moving beyond ruxolitinib failure in myelofibrosis: evolving strategies for second line therapy. Expert Opin Pharmacother. 2023;24:1091-1100.
- Harrison CN, Schaap N, Vannucchi AM, et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): a single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017;4:e317-e324.
- Harrison CN, Schaap N, Vannucchi AM, et al. Fedratinib in patients with myelofibrosis previously treated with ruxolitinib: an updated analysis of the JAKARTA2 study using stringent criteria for ruxolitinib failure. Am J Hematol. 2020;95:594-603.
- Mascarenhas J, Hoffman R, Talpaz M, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: a randomized clinical trial. JAMA Oncol. 2018;4:652-659.
- Gerds AT, Savona MR, Scott BL, et al. Determining the recommended dose of pacritinib: results from the PAC203 dose-finding trial in advanced myelofibrosis. Blood Adv. 2020;4:5825-5835.
- Mesa RA, Kiladjian JJ, Catalano JV, et al. SIMPLIFY-1: A phase III randomized trial of momelotinib versus ruxolitinib in janus kinase inhibitor-naive patients with myelofibrosis. J Clin Oncol. 2017;35:3844-3850.
- Harrison CN, Vannucchi AM, Platzbecker U, et al. Momelotinib versus best available therapy in patients with myelofibrosis previously treated with ruxolitinib (SIMPLIFY 2): a randomised, open-label, phase 3 trial. Lancet Haematol. 2018;5:e73-e81.
- Verstovsek S, Gerds AT, Vannucchi AM, et al. Momelotinib versus danazol in symptomatic patients with anaemia and myelofibrosis (MOMENTUM): results from an international, double-blind, randomised, controlled, phase 3 study. Lancet. 2023;401:269-280.Errata in: Lancet. 2023;401:1426; Lancet. 2023;402:2196.
- Ojjaara (momelotinib) tablets for oral use [prescribing information]. Durham, NC: GlaxoSmithKline; Septmber 2023.
- Tefferi A. Jaktinib and momelotinib for the treatment of myelofibrosis-birds of a feather? Am J Hematol. 2023;98:1517-1519.
- GSK. US Medical Affairs. Discontinuation and tapering of Ojjaara in phase 3 clinical trials. Accessed February 5, 2025. https://gskusmedicalaffairs.com/docviewer.html?cmd=GSKMedicalInformation&medcommid=MED--US-11592&token=23108-1da2884a-f34c-4d2e-bfe0-9d014c1c0c47&dns=gsk-medcomms.veevavault.com