Transplant-associated thrombotic microangiopathy (TA-TMA), an endothelial injury syndrome with incidence rates between 10% and 25% of transplant recipients, is a severe complication of hematopoietic stem-cell transplantation (HSCT) that has recently gained greater recognition in the posttransplant setting.1 TA-TMA is more frequently diagnosed after allogeneic HSCT than after autologous HSCT.2
The pathophysiologic mechanisms of TA-TMA are complex, encompassing injury-induced endothelial activation, the production of a procoagulant state, and immunologic responses that culminate in aberrant complement cascade activation and disseminated microthrombi formation.2-6
With complement dysregulation being the pathophysiologic nexus of TA-TMA, several studies have demonstrated a clinical response to eculizumab, a humanized monoclonal antibody that targets the terminal complement pathway by inhibiting C5 cleavage into C5a and C5b and ultimately preventing the formation of the terminal C5b-C9 complement complex.3,7,8
Ravulizumab was derived from eculizumab through a 4–amino acid substitution in eculizumab’s complementary binding and Fc regions, effectively augmenting C5 endosomal dissociation and ravulizumab’s recycling to the vascular compartment, which ultimately results in a longer duration of complete terminal complement inhibition.9
Ravulizumab is being increasingly used in the treatment of other complement-mediated disorders, such as primary atypical hemolytic uremic syndrome, and has demonstrated noninferiority versus eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria; however, ravulizumab is still under investigation for the treatment of TA-TMA.10-12 A study evaluating ravulizumab’s efficacy, safety, pharmacokinetics, and pharmacodynamics in adults and adolescents with HSCT-associated TA-TMA is currently underway.12,13
We present the case of an infant with relapsed or refractory infantile B-cell acute lymphoblastic leukemia (B-ALL) who received allogeneic HSCT, with subsequent development of TA-TMA, who was treated successfully with ravulizumab.
Case Report
A Black female patient presented at Nemours Children’s Hospital in Wilmington, Delaware, with infantile B-ALL (central nervous system [CNS] 2, KMT2a rearrangement t9;11) that was diagnosed at 4 months of age. She completed induction therapy per the Children’s Oncology Group’s AALL15P1 clinical trial,14 but early in consolidation therapy she had recurrent CNS disease with rising minimal residual disease that was consistent with relapsed or refractory B-ALL. The patient then received salvage therapy with CD19-directed chimeric antigen receptor T-cells and achieved complete remission, followed 60 days later by a 10/10 matched sibling allogeneic bone marrow transplant.
Of note, her donor had sickle-cell trait. Myeloablative conditioning included fludarabine, busulfan, and thiotepa with tacrolimus and methotrexate for graft-versus-host disease (GVHD) prophylaxis.
On day 31 after the transplant, the patient had worsening tachypnea and tachycardia, and a computed tomography scan of the chest, abdomen, and pelvis revealed the presence of a large pericardial effusion. She required a transfer to the cardiac intensive care unit, where she underwent emergent pericardiocentesis with a pericardial window. By day 41 after the transplant, she had increasing platelet and red blood cell (RBC) transfusion requirements. In the setting of ongoing and worsening serositis, which was noted through a reaccumulation of the pericardial effusion as well as pleural effusions, there were heightened concerns for possible TA-TMA because of its association with serositis.
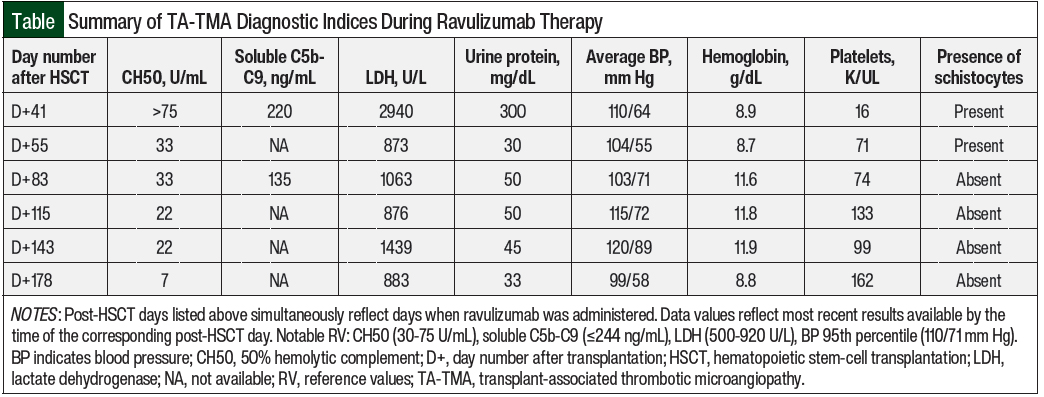
Laboratory studies at this time revealed a 50% hemolytic complement (CH50) level of >75 U/mL (normal, 30-75 U/mL), lactate dehydrogenase 2940 U/L (normal, 500-920 U/L), hemoglobin 8.9 g/dL, platelets 16 K/UL, urine protein 300 mg/dL, and the presence of schistocytes on peripheral smear.
The patient’s soluble C5b-C9 and C3 complement levels were high-normal at 220 ng/mL (normal, ≤244 ng/mL) and 143 mg/dL (normal, 72-164 mg/dL), respectively. Of note, ADAMTS13 (a disintegrin and metalloprotease with thrombospondin-1–like motif, member 13; also known as von Willebrand factor–cleaving protease) activity was normal at 75%. In addition to her serositis, the patient had other signs of multiorgan dysfunction syndrome, including hematochezia, hematuria, and an ongoing need for positive pressure ventilation.
Our diagnosis of TA-TMA in this patient was guided by the clinical and laboratory criteria proposed by Jodele and colleagues, including lactate dehydrogenase above normal for age, schistocytes on peripheral blood smear, de novo thrombocytopenia and anemia with associated increased transfusion requirements, hypertension less than the 99th percentile for age if aged <18 years, proteinuria ≥30 mg/dL twice on random urinalysis, and/or evidence of terminal complement activation via elevations in soluble plasma C5b-C9.7
High-risk TA-TMA requiring complement-inhibiting therapy is diagnosed when 5 of 7 diagnostic markers are present (with proteinuria and elevated soluble C5b-C9 being mandatory) or when 5 of 7 diagnostic markers are present with multiorgan dysfunction syndrome. With 5 of the 7 clinical criteria met for TA-TMA (elevated lactate dehydrogenase, schistocytes, increased platelet requirement, increased RBC requirement, proteinuria) in the setting of multiorgan dysfunction syndrome, a diagnosis of TA-TMA was made.
The only 2 criteria that were not met were severe hypertension (her blood pressure was less than the 99th percentile; however, it had risen to above the 95th percentile for age at the time of diagnosis, despite the administration of amlodipine 0.2 mg/kg twice daily) and elevated soluble C5b-C9.
At our institution, based on the preference of the nephrology team for ravulizumab over eculizumab for the treatment of atypical hemolytic uremic syndrome, ravulizumab was on formulary and a supply was readily available, whereas this was not the case for eculizumab. Given the patient’s multiorgan dysfunction syndrome, treatment with ravulizumab was initiated rather than wait to obtain eculizumab. Furthermore, given the association of calcineurin inhibitors with TA-TMA, her tacrolimus was weaned from a dose of 0.05 mg/kg enterally every 12 hours to completely finished by 10% increments every 3 to 7 days over a period of 6 weeks.
The patient’s treatment regimen included the administration of a 600-mg loading dose of ravulizumab intravenously over 90 minutes on day 41 after transplant, followed 2 weeks later by the initiation of a maintenance regimen of 600 mg intravenously over 60 to 120 minutes every 4 weeks. Each medication component of ravulizumab 100 mg/mL possessed a base component of 0.9% sodium chloride 6 mL to attain a final infusion concentration of 50 mg/mL, which specified a minimum total infusion time of 0.8 hours for the loading and maintenance doses in our patient, whose weight fell within the administration-dependent range of 10 to 20 kg.
The patient’s therapeutic goals included the clinical resolution of TA-TMA stigmata, as well as the adequate suppression of CH50 levels. The Figure details her clinical timeline and therapeutic response to ravulizumab through CH50 monitoring, whereas the Table summarizes the TA-TMA–related diagnostic indices that were recorded while she was receiving treatment.
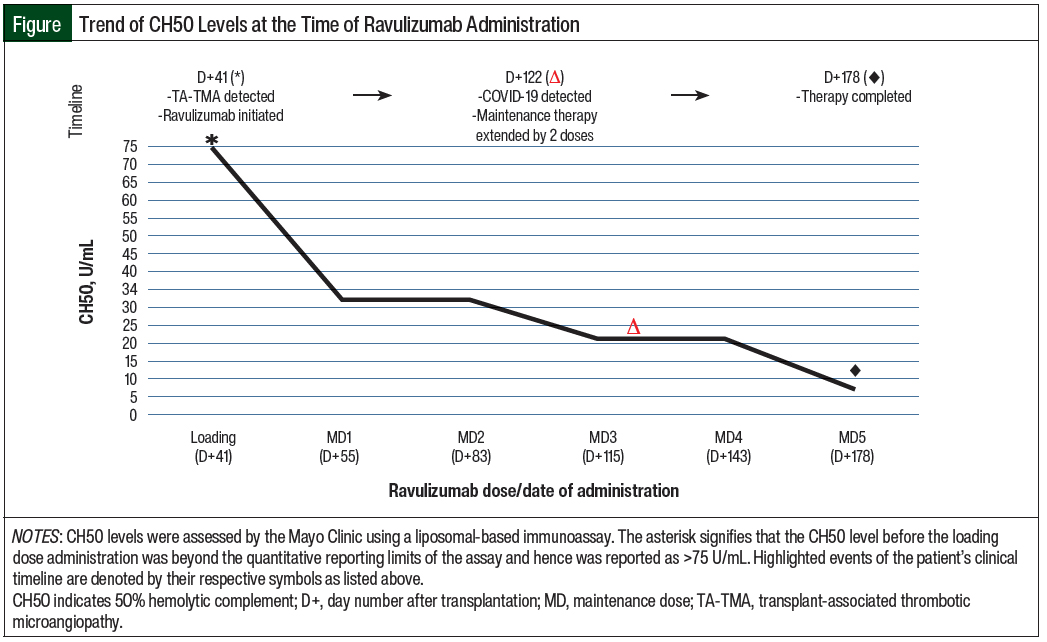
Clinical normalization was achieved approximately 2 weeks after the administration of the loading dose, which was marked by the resolution of her pericardial effusion and pleural effusions, the resolution of hypoxia without respiratory support, and the cessation of her aforementioned gastrointestinal symptoms with the resumption of enteral nutrition. Of note, endoscopic and pathologic evaluations of the patient’s gastrointestinal tract were obtained to evaluate for GVHD, which was negative (although no immunohistochemical staining for complement components related to TA-TMA was performed on the obtained biopsy samples).
Laboratory normalization of CH50 (to 33 U/mL) was also noted 2 weeks after the administration of the loading dose of ravulizumab, and her next repeat soluble C5b-C9 testing 2 weeks later showed marked improvement (from 220 ng/mL to 135 ng/mL).
A total of 5 maintenance doses of ravulizumab were administered approximately every 4 weeks, with CH50 levels ranging from <3 U/mL to 33 U/mL during that time.
By the time of maintenance dose 1, compared with the 2 weeks leading up to the loading dose administration, the patient’s RBC and platelet transfusion requirements decreased from a frequency of every 1 to 2 days to an average of every 3 days. Schistocytes were absent on peripheral smear by maintenance dose 2.
The patient’s proteinuria decreased from 300 mg/dL to 30 mg/dL after the loading dose was administered, and future measurements ranged between 33 mg/dL and 50 mg/dL, although the resolution of proteinuria was never confirmed on the completion of her TA-TMA therapy.
Her hypertension briefly improved, with her average blood pressure decreasing from 110/64 mm Hg to 104/55 mm Hg between the loading dose and maintenance dose 1 of ravulizumab. However, before maintenance dose 2 of ravulizumab, the patient had cryptogenic organizing pneumonia for which she received corticosteroids, which worsened her hypertension. With steroid tapering and the administration of amlodipine suspension 0.25 mg/kg 2 times daily, her average blood pressure gradually improved by maintenance dose 5 to 99/58 mm Hg, which was below the 95th percentile for age and height. Her lactate dehydrogenase quickly normalized to 873 U/L (institutional reference range: 500-920 U/L and remained unremarkable until maintenance dose 4, when it was markedly elevated at 1439 U/L.
Notably, 3 weeks before receiving dose 4 of ravulizumab, the patient tested positive for COVID-19 and had mild symptoms. Her lactate dehydrogenase was persistently elevated and ranged between 977 U/L and 1602 U/L in the 4 weeks after maintenance dose 4 was administered.
The original plan was to administer 3 maintenance doses of ravulizumab. However, because we were concerned that the patient’s acute COVID-19 infection could exacerbate TA-TMA given its inflammatory nature, and because she had already tolerated previous doses of ravulizumab well, 2 additional maintenance doses were administered, for a total of 5 doses of ravulizumab.
Because of the increased risk for meningococcal infection with complement inhibition, the patient also received amoxicillin suspension 10 mg/kg every 12 hours for prophylaxis during treatment and up to 3 months after her final dose of ravulizumab.
At the time of this writing (approximately 1 year after transplant), it has been more than 6 months since the patient received her last dose of ravulizumab, and her leukemia and TA-TMA remain in remission.
Discussion
TA-TMA is an increasingly recognized and serious complication of HSCT, with a reported nonrelapse mortality rate of 43.6% in patients with TA-TMA versus 7.8% in those without TA-TMA at 1 year after transplantation.4 The high mortality rate should promote further investigation and the application of new and existing therapies for TA-TMA.
To date, eculizumab is the standard for targeted anticomplement therapy in patients with TA-TMA. In contrast to patients with atypical hemolytic uremic syndrome, who will require lifelong treatment with a complement inhibitor, eculizumab treatment can ultimately be discontinued in patients with TA-TMA after successful therapy is completed. However, achieving a response in patients with TA-TMA requires higher and more frequent dosing of eculizumab in comparison with atypical hemolytic uremic syndrome, and some patients who are unable to reach therapeutic levels of eculizumab have died secondary to TA-TMA.5,15
In this report, we describe a patient who received ravulizumab instead of eculizumab as a result of availability. Approximately 2 weeks after the patient received the loading dose of ravulizumab, she achieved clinical normalization with the resolution of symptoms, an improvement in laboratory results, decreased transfusion requirements, improved hypertension, and the disappearance of schistocytes. Because endothelial damage is an initiating event in TA-TMA, it is worth mentioning that the donor had sickle-cell trait, although it is unknown if that contributed to this case.
The efficacy of complement inhibition can be measured using CH50 levels. Whereas for eculizumab there are existing data for expected CH50 levels during treatment and how dosing should be modified based on these levels, it is currently unknown what CH50 levels should be for ravulizumab at what time points and how dosing should be adjusted if CH50 levels are not adequately suppressed.
Moreover, in contrast to the published CH50 goals of ≤3 when measured with a standard enzyme immunoassay or ≤15 for the Diamedix hemolytic assay,5 this patient’s CH50 levels were measured at the Mayo Clinic using a liposomal-based immunoassay in which reference levels for CH50 are different from these other assays.
Regardless, symptomatic and laboratory improvements corresponded with a decrease in CH50 levels that were below normal, and overall complement suppression was demonstrated with ravulizumab.
Although they share a highly comparable mechanism of action, the primary benefit of ravulizumab over eculizumab is a much longer half-life (51.8 days for ravulizumab in adults and pediatric patients vs approximately 11-17 days for eculizumab).16,17 This longer half-life has multiple benefits.
For patients requiring courses that extend into the outpatient setting, patient satisfaction and preference strongly favor ravulizumab over eculizumab.18 A study of adults with paroxysmal nocturnal hemoglobinuria that compared the use of eculizumab with ravulizumab showed that ravulizumab led to more sustained complement inhibition, which reduced breakthrough symptoms.19
Moreover, less frequent dosing of ravulizumab versus eculizumab in patients with paroxysmal nocturnal hemoglobinuria (every 8 weeks vs every 2 weeks, respectively) led to a cost-savings of 10%, resulting in more than $1 million in cost-savings per quality-adjusted life-year gained.19,20
For patients with atypical hemolytic uremic syndrome, the reduced frequency of dosing for ravulizumab over eculizumab reduced costs by more than 30%, resulting in a total cost-savings of more than $5.8 million.21
Conclusion
This case demonstrates that ravulizumab treatment in a patient with TA-TMA can lead to a favorable clinical response. In situations where eculizumab is not readily available, the use of ravulizumab as an alternative treatment could be considered. However, we cannot extrapolate from our single patient’s response how others might respond.
We acknowledge that the unique diagnosis of infantile B-ALL and the distinct physiology that our infant patient possesses merit distinguishment from patients in other age groups. Nonetheless, studies that evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of ravulizumab in adults and adolescents with HSCT-associated TA-TMA are currently being conducted.
Author Disclosure Statement
Dr Gonzales, Dr White, Dr Davidow, Dr Mangum, and Dr Grimm have no conflicts of interest to report.
References
- Laskin BL, Goebel J, Davies SM, Jodele S. Small vessels, big trouble in the kidneys and beyond: hematopoietic stem cell transplantation–associated thrombotic microangiopathy. Blood. 2011;118:1452-1462.
- Chapin J, Shore T, Forsberg P, et al. Hematopoietic transplant-associated thrombotic microangiopathy: case report and review of diagnosis and treatments. Clin Adv Hematol Oncol. 2014;12:565-573.
- Dvorak CC, Higham C, Shimano KA. Transplant-associated thrombotic microangiopathy in pediatric hematopoietic cell transplant recipients: a practical approach to diagnosis and management. Front Pediatr. 2019;7:133. doi: 10.3389/fped.2019.00133.
- Jodele S, Davies SM, Lane A, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014;124:645-653.
- Jodele S, Laskin BL, Dandoy CE, et al. A new paradigm: diagnosis and management of HSCT-associated thrombotic microangiopathy as multi-system endothelial injury. Blood Rev. 2015;29:191-204.
- Young JA, Pallas CR, Knovich MA. Transplant-associated thrombotic microangiopathy: theoretical considerations and a practical approach to an unrefined diagnosis. Bone Marrow Transplant. 2021;56:1805-1817.
- Jodele S, Dandoy CE, Lane A, et al. Complement blockade for TA-TMA: lessons learned from a large pediatric cohort treated with eculizumab. Blood. 2020;135:1049-1057.
- Zhang R, Zhou M, Qi J, et al. Efficacy and safety of eculizumab in the treatment of transplant-associated thrombotic microangiopathy: a systematic review and meta-analysis. Front Immunol. 2021;11:564647. doi: 10.3389/fimmu.2020.564647.
- Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5-inhibitor–experienced adult patients with PNH: the 302 study. Blood. 2019;133:540-549.
- Barbour T, Scully M, Ariceta G, et al. Long-term efficacy and safety of the long-acting complement C5 inhibitor ravulizumab for the treatment of atypical hemolytic uremic syndrome in adults. Kidney Int Rep. 2021;6:1603-1613.
- Lee JW, Sicre de Fontbrune F, Wong Lee Lee L, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood. 2019;133:530-539.
- Ravulizumab in thrombotic microangiopathy after hematopoietic stem cell transplant. NLM identifier: NCT04543591. Updated January 27, 2023. https://clinicaltrials.gov/ct2/show/NCT04543591. Accessed February 24, 2023.
- Study of Ravulizumab in Pediatric Participants With HSCT-TMA. NLM identifier: NCT04557735. Updated February 1, 2023. https://clinicaltrials.gov/ct2/show/NCT04557735. Accessed March 5, 2023.
- Children’s Oncology Group. AALL15P1: a groupwide pilot study to test the tolerability and biologic activity of the addition of azacitidine to chemotherapy in infants with acute lymphoblastic leukemia (ALL) and KMT2A (MLL) gene rearrangement. https://childrensoncologygroup.org/aall15p1. Accessed February 24, 2023.
- Jodele S, Fukuda T, Vinks A, et al. Eculizumab therapy in children with severe hematopoietic stem cell transplantation–associated thrombotic microangiopathy. Biol Blood Marrow Transplant. 2014;20:518-525.
- Soliris (eculizumab) injection, for intravenous use [prescribing information]. Alexion Pharmaceuticals; November 2020. https://alexion.com/Documents/Soliris_USPI.pdf. Accessed February 24, 2023.
- Ultomiris (ravulizumab-cwvz) injection, for intravenous or subcutaneous use [prescribing information]. Alexion Pharmaceuticals; July 2022. https://alexion.com/Documents/Ultomiris_USPI.pdf. Accessed February 24, 2023.
- Peipert JD, Kulasekararaj AG, Gaya A, et al. Patient preferences and quality of life implications of ravulizumab (every 8 weeks) and eculizumab (every 2 weeks) for the treatment of paroxysmal nocturnal hemoglobinuria. PLoS One. 2020;15:e0237497. doi: 10.1371/journal.pone.0237497.
- O’Connell T, Buessing M, Johnson S, et al. Cost-utility analysis of ravulizumab compared with eculizumab in adult patients with paroxysmal nocturnal hemoglobinuria. Pharmacoeconomics. 2020;38:981-994.
- Brodsky RA, Peffault de Latour R, Rottinghaus ST, et al. Characterization of breakthrough hemolysis events observed in the phase III randomized studies of ravulizumab versus eculizumab in adults with paroxysmal nocturnal hemoglobinuria. Haematologica. 2021;106:230-237.
- Wang Y, Johnston K, Popoff E, et al. A US cost-minimization model comparing ravulizumab versus eculizumab for the treatment of atypical hemolytic uremic syndrome. J Med Econ. 2020;23:1503-1515.